Research Article
European Clinical Laboratory, Molecular and Pathological (ECMP) criteria for prefibrotic JAK2V617F-Thrombocythemia and Polycythemia Vera versus MPL515- and CALR-Thrombocythemia and Myelofibrosis: From Dameshek to Michiels 1950-2018

Jan Jacques Michiels1,2,5*, Zwi Berneman2, Wilfried Schroyens2, Fibo W J ten Kate3, King Lam3 and Hendrik De Raeve4,5
1Multidisciplinary Internist, Scientific Investigator, Department of Hematology & Blood Coagulation, Erasmus Free University Network, Europe, Rotterdam, Netherlands
2Hematologist, Internist, Department of Hematology, University Hospital, Antwerp, Belgium
3Pathologist, Department of Pathology, Erasmus University Medical Center Rotterdam, Netherlands
4Pathologist, Department of Pathology, OLV Hospital Aalst and Free University Hospital, Brussels, Belgium
5European Working Groups on Myeloproliferative Neoplasms (EWG.MPN 1998-2018), Good heart Institute & Foundation, Freedom of Science & Education Rotterdam, Netherlands
*Address for Correspondence: Jan Jacques Michiels, Multidisciplinary Internist & Scientific Investigator, European Working Group on Myeloproliferative Neoplasm EWG.MPN 1998-2018, Goodheart Institute in Nature Medicine, Rotterdam, Europe, Erasmus Tower, Veenmos 13, 3069 AT Rotterdam, Netherlands; Tel: +316-26970534; Email: [email protected]
Dates: Submitted: 18 February 2019; Approved: 28 February 2019; Published: 01 March 2019
How to cite this article: Michiels JJ, Berneman Z, Schroyens W, J ten Kate FW, Lam K, et al. European Clinical Laboratory, Molecular and Pathological (ECMP) criteria for prefibrotic JAK2V617F-Thrombocythemia and Polycythemia Vera versus MPL515- and CALR-Thrombocythemia and Myelofibrosis: From Dameshek to Michiels 1950-2018. Int J Bone Marrow Res. 2019; 2: 001-017. DOI: 10.29328/journal.ijbmr.1001002
Copyright License: © 2019 Michiels JJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Abstract
The broad spectrum of heterozygous versus homozygous JAK2V617F mutated MPN consists ET, ET with early features of PV (prodromal PV), classical PV, masked PV, advanced PV and post-PV myelofibrosis. Combined use of bone marrow histology and increased erythrocyte counts above 5.8x1012/L can replace increased red cell mass at time of presentation as the pathognomonic clue for the correct diagnosis of hetero/homozygous or homozygous mutated PV. Erythrocyte counts are in the normal range below 5.8x1012/L in heterozygous JAK2V617F mutated ET and prodromal PV but above 5.8x1012/L in heterozygous-homozygous or homozygous mutated PV. The bone marrow cellularity and morphology in pre-fibrotic ET, prodromal PV and PV carrying the JAK2V617F mutation are overlapping showing clustered increase of large mature pleomorphic megakaryocytes (M) with no increase of cellularity (<60%) in ET. The bone marrow is hypercellular (60%-80%) due to increased erythropoiesis megakaryopoiesis (EM) in prodromal and classical PV and trilinear hypercellular (80%-100% due increased megakaryopoiesis, erythropoiesis and granulopoiesis (EMG) in advanced PV and masked PV. Bone marrow cellularity ranging from normal (<60%) in ET to increased erythropoiesis (EM) in prodromal PV to hypercellular (80-100%) in advanced PV and masked PV largely depends on increasing JAK2V617F mutation load from low to high on top of other biological MPN variables like constitutional symptoms during long-term follow-up. MPL515 mutated ET is featured by an increase of clustered small and giant megakaryocytes with hyper-lobulated staghorn-like nuclei in a normal cellular bone marrow. The third entity of pronounced JAK2/MPL wild type ET associated with primary megakaryocytic granulocytic myeloproliferation (PMGM) without PV features proved to be caused by calreticulin (CALR) mutation. CALR mutated thrombocythemia is characterized by dual proliferation of megakaryocytic and granulocytic bone marrow proliferation of dense clustered large to giant immature dysmorphic megakaryocytes with bulky (bulbous) hyperchromatic nuclei, which are not seen in MPL515-mutated Thrombocythemia and JAK2V617F-Thrombocythemia, prodromal PV and classical PV.
Introduction
Focusing since 1975 on the discovery and elucidation of erythromelalgia caused by platelet-mediated inflammatory and thrombosis in thrombocythemia in ET and PV patients [1-3], and on the association of migraine-like microvascular cerebral transient ischemic attacks (MIA) as specific presenting symptoms of ET [4], we were able to document the early pre-fibrotic stages of ET and PV by the combined use of clinical, laboratory and bone marrow histopathology features (Figures 1-5, Tables 1,2) [4,5]. As no definition was available for early stage ET at that time, we prospectively performed clinical and basic research studies on the causal relationship between aspirin-sensitive erythromelalgia and atypical MIA in thrombocythemia of ET and PV patients in the period of 1975 and 1998. Between 2000 and 2008 we performed prospective studies on the clinical and pathobiological features of pre-fibrotic JAK2V617F mutated ET and PV and JAK2 wild type ET patients.
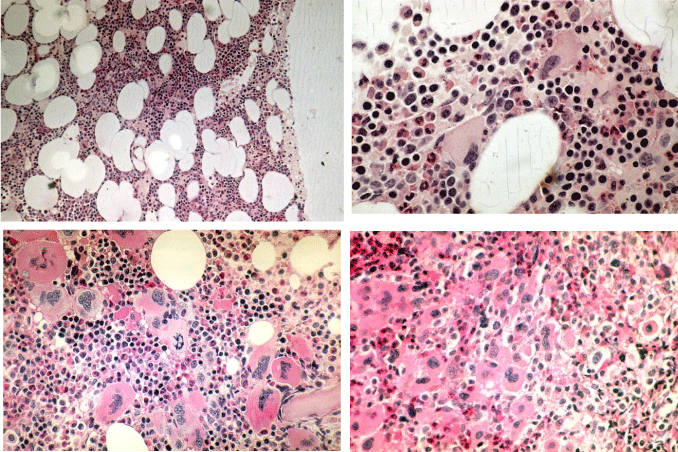
Figure 1: ET (upper) and PV (lower) bone marrow features have similar pleiomorphic megakaryocytes. increase of clustered enlarged megakaryocytes in a normocelluar ET bone marrow with stainable iron [5,6,13,16,34]. Local increase of erythropoiesis (bold arrows) in areas of loose clustered pleiomorphic megakaryoctyes in patients with essential thrombocythemia: ET bone marrow picture. Dense clustered pleiomorphic megakaryocytes in PV/RF (right bottom) in advanced PV show dysmorphic nuclei [5,6,13].
Figure 1: Spectrum of ET and PV bone marrow features in 155 bone marrow biopsies with clustered enlarged “PV” megakaryocytes of PV patients from the PVSG 01 study ranging from an ET bone marrow picture (A left and middle B): arrows indicate the same area of clustered pleiomorphic megakaryocytes) to a classical PV bone marrow picture (A right) with trilinear eryhtro/megakaryo/granulocytic (EMG) hyperplasia. Source Ellis et al PVSG 1986 [11]. Courtesy of Wasserman ASH 1995. Wasserman LR, Berk PD, Berlin NI. Polycythemia Vera and the myeloproliferative disorders. 1995, WB Saunders ISBN 0-7216-4213-6. An ET picture was observed in 10 PV, an ET/PV bone marrow picture with increased cellularity (60-80%) was detected in 45 PV, and a hypercellular (80-100%) PV picture was recorded in 90 evaluable bone marrow biopsies of 155 PV patients, who had a documented increased red cell mass in the PVSG 01 study [11].
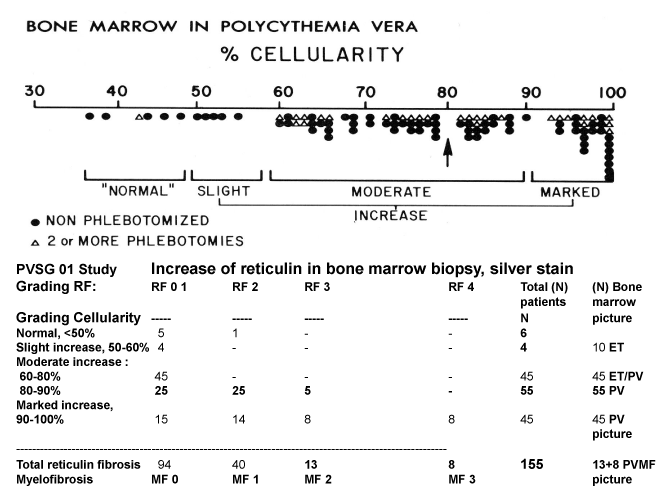
Figure 3: Grading of bone marrow cellularity and reticulin fibrosis in the PVGS-01 study of 155 PV patients [11].
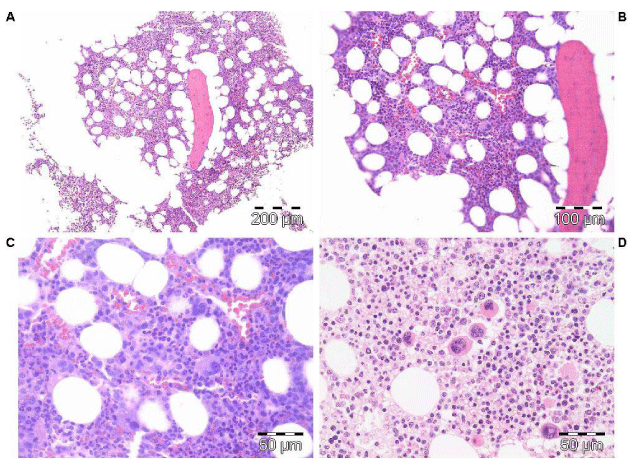
Figure 4: Clinical history and bone marrow histology in Case 8 in table 6 with prefibrotic JAK2V617F positive PV and a diagnostic ET/PV (forme fruste PV) bone marrow picture.
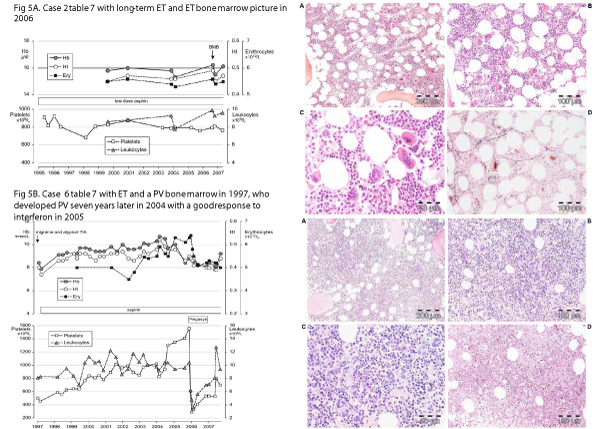
Figure 5: A: Clinical history and bone marrow histology in Case 6 table 6 with JAK2V617F positive ET with a typical PV bone marrow in 1997, who subsequently developed classical PV seven years later in 2004 with a good response to interferon in 2005. B: Clinical history and bone marrow histology in Case 6 table 6 with JAK2V617F positive ET with a typical PV bone marrow in 1997, who subsequently developed classical PV seven years later in 2004 with a good response to interferon in 2005.
| Table 1: The 1980 RCP criteria of essential thrombocythemia; ET [2-5]. |
| Major criteria |
| A1 Persistent platelet count in excess of 400x109/. |
| A2 Increase and clustering of enlarged mature megakaryocytes in bone marrow biopsy |
| Confirmative criteria |
| B1 Presence of large platelets in a peripheral blood smear |
| B2 Absence of any underlying disease for reactive thrombocytosis and normal ESR. |
| B3 No or slight splenomegaly on palpation or scan (<15 cm) |
| B4 Increase of LAF-score and no signs of fever or inflammation |
| Exclusion criterion |
| Ph+ chromosome and any other cytogenetic abnormality in blood or bone marrow cells |
| The 1980 RCP criteria for PV [11,12,22] on top of the 1975 PVSG criteria for PV [10,14] |
| Major |
| A1 Raised red cell mass. Male >36 ml/kg, female >32 ml/kg10 consistent with erythrocytes count of >6x1012/L (Dameshek 1940 [22], Michiels, Figure 1) |
| A2 Absence of primary or secondary erythrocytosis by clinical and laboratory tests. |
| A3 Slight, moderate or marked increase in bone marrow biopsy material of: clustered, mature, enlarged pleiomorphic megakaryocytes with hyperlobulated nuclei and moderate to marked increase cellularity of megakaryopoiesis and erythropoiesis or typically trilinear mega-erythro-granulopoiesis (EMG). |
| A typical PV bone marrow excludes erythrocytosis [12]. |
| No or presence of reticuline fibers and no collagen fibers (no dry tap) |
| Minor |
| B1 Thrombocythemia, persistent increase of platelet >400x109/ |
| B2 Leukocytosis, leucocyte count >10x/L and low erythrocyte sedimentation rate (ESR) |
| B3 Raised leukocyte alkaline phosphatase score >100, absence of fever or infection |
| B4 Splenomegaly on palpation or on isotope/ultrasound scanning |
| A1+ A3 plus one of B establishes PV and excludes any variant of erythrocytosis. |
| Grading of bone marrow cellularity according to Kurnick 197211 and 1980 RCP [2-5]. |
| N: normal cellularity (<50-60%) |
| +: increased cellularity as judged by decease fat cells (cellularity 60-80%) |
| ++: hypercellular with absence of fat cells (80-100%) |
| Table 2: Grading of reticulin and collagen fibrosis by Ellis (PVSG 1986) [11], Georgii (1990, 1996) [26,45], Michiels (2005) [6,16] and Thiele (2005) [6,17]. | ||
| Grading reticulin fibrosis PVSG [11] |
Grading MF ECP criteria |
Description of reticulin and collagen myelofibrosis (MF) as a secondary event in myeloproliferative disorders (MPD) |
| Normal (N) RF-0 |
N MF 0 | No reticulin fibers, occasional individual fibers or focal areas with tiny amount of reticulin fiber network |
| Slight increase RF 1 |
RF + MF 0 | Fine reticulin fiber network throughout much of section and no course reticulin fibers |
| Moderate increase RF 2 |
RF ++ MF 1 | Diffuse fine reticuline network with focal collections of thick course reticulin fibers and no collagenization |
| Marked increase BM dry tap RF 3 = RCF |
RF +++ MF 2 | Diffuse and dense increase in reticulin with extensive intersections, and presence of collagen fibers and no osteosclerosis |
| OS Dry tap RF 4 |
ScleroticMF 3 | Diffuse and dense reticulin with with coarse bundles of collagen associated with significant osteosclerosis |
| Translation of RF and RCF grading into grading of myelofibrosis (MF) according to Michiels & Thiele 2005 [6,16]. | ||
During the years, ET and PV patients were diagnosed according to the Rotterdam Clinical and Pathologic (1980 RCP) [1-5], the European Clinical and Pathologic (2002-2005 ECP [6] http://www.mpn-stichting.nl/doctors_brochure_2004.pdf) and the European Clinical Molecular and Pathological (2007 ECMP) [7], criteria for the classification of pre-fibrotic ET and PV and ET with a hypercellular megakaryocytic granulocytic (MG) bone marrow histology. In this contribution, we present the updated results of our prospective studies in view of a critical approval of the literature. This report provide good evidence that the characteristic features of pre-fibrotic myeloproliferative neoplasms (MPN) can be subclassified as JAK2V617F mutated ET, ET with early features of PV (prodromal PV), classical PV, and ET with hypercellular megakaryocytic granulocytic proliferation as the three main JAK2V617F mutated variants of pre-fibrotic MPN (Figures 1 to 10) [1-48]. According to Dameshek (1950) [23], PV is a trilinear myeloproliferative disease (MPD) caused by the JAK2V617F mutation (Vainchenker and his team 2005, Figure 6) [32,33,36]. According to Dameshek (1951), PV is clearly distinct from megakaryocytic leukemia (ML) [1,9] or primary thrombocythemia hemorrhagica (PTH) without features of PV (Laszlo 1975) [8]. Here we demonstrate that ML or PTH belong to the JAK2 wild type variants of pre-fibrotic ET carrying one of the MPL [42,43] or CALR [44] mutations. JAK2 wild type hypercellular ET associated with primary megakaryocytic granulocytic myeloproliferation (PMGM) [26,45,46}, proved to be CALR mutated [44,47,48], as the third distinct MPN when the Clinical Laboratory Molecular and Pathologic (CLMP) criteria are applied (Tables 3-5) [49,50].
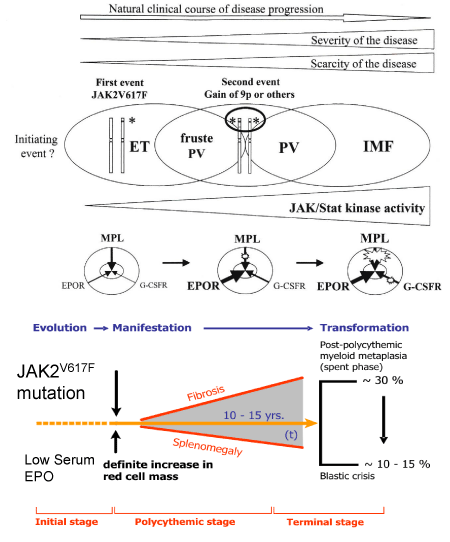
Figure 6: Upper part. Disease heterogeneity of JAK2V617F myeloproliferative disease according to Villeval, James, Pisani, Casedevall & Vainchenker 2006 [36]. The level and duration of JAK2V617F expression directly contribute to the diversity of trilinear myeloproliferative neoplasms. The JAK2V617F occurs in a hematopoietic stem cell and give rise to the onset of ET and PV. The trilinear MPN progresses from heterozygous ET to hetero/homozygous ET/PV (forme fruste PV) and homozygous JAK2V6167F mutated classical PV through mitotic recombination at chromosome 9p resulting in the loss of heterogeneity (9pLOH)36. A low level of heterozygous JAK2V617F kinase activity is enough to activate the MPL receptor to produce the clinical ET phenotype. A higher level of hetero/homozygous and a high level of homozygous JAK2V617F kinase activity is needed to activate the EPOR, MPL and G-CSFR receptors to produce an ET/PV (forme fruste PV), overt classical PV and advanced masked PV [32,33,36]. Lower part. Dynamics of the JAK2V617F mutated trilinear MPN disease including the sequential evolution of ET, forme fruste PV (prodromal PV), classical PV and advanced PV complicated by post-PV myeloid metaplasia of the spleen and secondary myelofibrosis according to ECP and ECMP classifications of the Myeloproliferative Neoplasms (MPN) designed by Michiels & Thiele [16,34,35,47-50].
| Table 3: 2018 Clinical, Laboratory, Molecular and Pathobiological (CLMP) criteria for diagnosis of JAK2V617F mutated trilinear myeloproliferative neoplasms essential thrombocythemia (ET) and polyvythemia vera (PV) according to De Raeve et al. [51,52] | |
| Clinical and molecular (CLM) criteria | Bone marrow pathology (P) criteria |
| Prefibrotic ET | Normocellular ET |
|
Normocellular bone marrow (<60%), Megakaryocytic (M) proliferation of clustered of medium sized to large (pleomorphic) mature megakaryocytes in a normocellular bone marrow (<60%), no proliferation of erythropoiesis and granulopoiesis. Reticuline fibrosis (RF) 0 or 1 |
| Prefibrotic prodromal PV | ET with bone marrow features of PV |
|
Increased cellularity (60-80%) due to increased erythrocytic, megakaryocytic (EM) proliferation or trilinear erythrocytic, megakaryocytic, granulocytic (EMG) proliferation. Increase of clustered medium sized to large (pleomorphic) mature megakaryocytes. RF 0 or 1 |
| Prefibrotic hypercellular PV | Classical PV, Masked PV, Advanced PV |
| A 1. Erythrocytes >5.8x1012/L males and >5.6x1012/L females. Hemoglobin and Hematocrit upper range of normal or increased A 2. Heterozygous/homozygous JAK2V617F mutation load around and above 50% A 3. Low serum Epo, increased LAP B 1. Persistent increase of platelet count x109/: grade I: 400-1000, grade II: >1000. B 2. Granulocytes >8 x109/l or Leukocytes >10 x109/l and raised LAP-score or increased CD11b expression in the absence of fever or infection B 3. Myeloid neoplasia of the spleen à splenomegaly on ultrasound echogram (>12 cm length in diameter) or on palpation. B 4. Spontaneous endogenous erythroid colony (EEC) formation (optional) A = major and B = minor criteria for PV |
PV. Increased cellularity (60-100%) due to increased erythrocytic, megakaryocytic (EM) proliferation or trilinear erythrocytic, megakaryocytic and granulocytic (EMG) proliferation. Increase of clustered medium to large (pleomorphic) megakaryocytes with hyperlobulated nuclei. Absence of stainable iron. Masked PV. Hypercellular ET due to increased erythrocytic, megakaryocytic and granulocytic (prefibrotic EMG) bone marrow proliferation or increased megakaryocytic, granulocytic myeloproliferation (MG fibrotic stage) with relative reduced erythroid precursors and presence of immature pleiomorphic megakaryocytes with hyperploid or clumpsy nuclei. Advanced PV: stage 1: Hb between <14 g/dL and >12 g/dl, normal LDH and CD34+ . stage 2: anemia Hb <12 to >10 g/dL, LDH↑, and splenomegaly. stage 3: severe anemia, Hb <10 g/dL,LDH↑↑, CD34+ , leukoerythroblastose, tear drop erythrocytes, and large spleen Grading of reticulin fibrosis (PVSG) and MF Table 2 (ECP) |
| Table 4: 2018 Clinical Laboratory, Molecular and Pathological (CLMP) criteria for the diagnosis of normocelular monolinear megakaryocytic (M) bone marrow proliferation and essential thrombocythemia (ET) carrying one of the MPL515 mutations according to De Raeve et al. [51,52] | |
| CLM criteria MPL515 Thrombocythemia | Bone marrow pathology (P) |
|
Megakaryocytic (M) bone marrow proliferation in a normocellular (<60%) bone marrow featured by large to giant mature megakaryocyte with hyper-lobulated, staghorn-like nuclei. No increase of erythropoiesis, and granulopoiesis No or slight increase in reticulin RF 0/1 Grading of reticulin fibrosis (RF), collagen fibrosis (CF) and myelofibrosis (MF) The natural history of MPL515-Thrombocythemia and myelofibrosis occurs in a normocellular or hypocellular bone marrow and clearly differs from JAK2V617F- and CALR-thrombocythemia |
| Table 5: 2018 Clinical Laboratory, Molecular and Pathological (CLMP) criteria for hypercellular ET associated with primary megakaryocytic, granulocytic myeloproliferation (PMGM) or JAK2 wild type Thrombocythemia caused by calreticulin (CALR) mutations defined by Michiels & Thiele. | |
| CLM criteria CALR-Thrombocythemia A1 Megakaryocytic Leukemia (ML) according to Dameshek or Essential Thrombocythemia (ET) without features of PV and no preceding or allied other subtype of myeloproliferative neoplasm PV, CML or MDS. The main presenting features is pronounced isolated thrombocythemia with platelet count around or above 1000x109/L A2 CALR mutation and JAK2 wild type Clinical stages (C) of CALR Thrombocythemia C 1. Early clinical stage: Hb >12g/dL, slight to moderate splenomegaly, thrombocytosis around or above 1000x109/L, normal LAP score C2. Intermediate clinical stage: slight anemia Hb <12 to >10 g/dL, decreasing platelet count, splenomegaly, increased LDH and definitive tear drop erythrocytes C3. Advanced stage: anemia Hb <10 g/dL, tear drop erythrocytes, increased LDH, increased CD34+ cells, pronounced splenomegaly, normal or decreased platelet counts, leucocytosis or leukopenia. |
Pathological (P) criteria of CALR MPN ML or ET due to mono-linear megakaryocytic (M) bone marrow proliferation of large to giant megakaryocytes with immature bulbous nuclei in a normocellular bone marrow Primary dual megakaryocytic granulocytic myeloproliferation (PMGM) in a hypercellular bone marrow with relative or absolute reduction of erythropoiesis and erythroid precursors. Abnormal dense clustering and increase in atypical medium sized, large to giant immature megakaryocytes containing bulbous cloud-like nuclei and definitive maturation defects No features of PV in blood and bone marrow MFGrading reticulin fibrosis (RF), myelofibrosis (MF) MF 0 Pre-fibrotic CALR MM, no reticulin fibrosis RF 0/1 MF 1 Early fibrotic CALR PMMG with increased RF 2 MF 2 Fibrotic CALR PMMG with increased RF grade 3 MF 3 Advanced CALR PMMG with fibrosis-osteosclerosis |
Methods
In1975 the minimum criterion for the diagnosis of ET proposed by the Polycythemia Vera Study Group (PVSG) was 1000x109/. ET at platelet count above 1000x109/ usually presented with the clinical picture of hemorrhagic thrombocythemia (HT) [8,9]. In view of our discovery of platelet-mediated erythromelalgia as a specific and early presenting symptoms of thrombocythemia in ET we were able to document the very early stage of ET at platelet counts between 400 and 1000x109/ by the use of the Rotterdam Clinical and Pathological (1980 RCP) criteria for ET (Table 1) [2,3]. The 1980 RCP criteria of ET were determined by careful prospective documentation of peripheral blood and bone marrow smears and bone marrow biopsy material [2+4]. Platelets in excess of 400x109/, and an increase of clustered enlarged megakaryocytes in a bone marrow biopsy material was found to be diagnostic for ET and excluded reactive thrombocytosis. On top of the clinical PVSG criteria for PV [8-10] we used since 1980 bone marrow histopathology and erythrocyte count above 6x1012/L to confirm PV and exclude all variants of primary and secondary erythrocytosis [11,12]. The 1980 RCP criteria for PV replaced O2-saturation of >92% by bone marrow biopsy, changed splenomegaly by bone marrow biopsy as a major criterion, and used splenomegaly as a minor criterion (Table 1) [1-3]. Raised B12 (>900 ng/L) or raised B12 binding capacity (>2200 ng/L) were skipped as completely irrelevant for the diagnosis of early stage PV. Raised score for leukocyte alkaline phosphatase (LAP) stain and normal erythrocyte sedimentation rate (ESR) are specific features of ET and PV [13,14]. Applying the 1980 RCP criteria we could detect masked and prodromal cases of ET and PV. In 2002 Michiels & Thiele proposed the European Clinical and Pathological (ECP, http://www.mpn-stichting.nl/doctors_brochure_2004.pdf) criteria for ET, PV and defined ET with a hypercellular bone marrow defined as PMGM [15-17]. In this report we extended the RCP and ECP (Table 1) into the 2006-2018 European Clinical, Molecular and Pathological (ECMP) for the diagnosis and classification of JAK2V617F mutated ET, prodromal PV, classical PV and masked PV as clearly distinct from MPL515 mutated ET and of JAK2/MPL wild type ET associated with CALR mutated PMGM (Tables 1,2) [13-16,34,46-50].
Bone marrow biopsies from the iliac crest were stained with hematoxylin and eosin for histopathology evaluation. All bone marrow biopsies were evaluated by expert pathologists for morphology, grading of cellularity and scoring of reticulin fibers using the silver impregnation stain according to PVSG and ECP recommendations (Ellis et al., Table 2) [11]. Additional screening was performed for endogenous erythroid colony (EEC), formation of nucleated peripheral blood or aspirated bone marrow cells, serum erythropoietin (EPO) levels [19-21] and JAK2V617F mutation using the PCR test according to Baxter et al. [18]. With the advent of JAK2V617F as the cause of trilinear MPN [32,33], MPL515 mutations [42,43] as another cause of ET and MF and JAK2/MPL wild type CALR mutated PMGM [26,34,44-46] we extended the ECMP criteria [47,48], into the Clinical Laboratory Molecular and Pathobiological (CLMP) features [49,50], for classification and staging three distinct JAK2V617F ET, PV and MF, JAK2 exon 12 PV versus MPL515 and CALR mutated thrombocythemia and myelofibrosis without features of PV (Tables 3-5) [46-50].
ET and PV result analysis using the RCP, ECMP and CLMP criteria
Between 1975 to 1985, we prospectively studied 30 consecutive thrombocythemia patients diagnosed as ET in 14 and PV in 16. The clinical features of erythromelalgia in ET and PV cases have been reported in 1985 [3]. Eleven of 14 ET patients had platelet counts below 1000x109/, in whom the diagnosis of ET would have been overlooked by the PVSG criteria at that time (1975-1985). Spleen size on scan was slightly increased in 5 of 14 ET and in 13 of 16 PV patients. Leukocyte count counts was increased (>10x109/) in 5 out of 14 patients with ET and in 14 of 16 PV patients. LAP score was increased (>100) in 12 out of 14 ET and in all PV patients. Increase of clustering of enlarged megakaryocytes and a normal erythropoiesis was present in bone marrow smears and biopsies of 14 ET and 16 PV patients. Increase of clustered pleomorphic megakaryocytes and no increase of cellularity (Figure 1) was seen in 7 of 14 ET and in one of 16 PV patients. Increase of bone marrow cellularity (1+ = 60-80%) due to increased erythropoiesis leading to an ET/PV picture was seen in 3 ET and 4 PV patients (Figure 1). Pronounced increase of cellularity (2+ =80-100%) due to trilinear erythrocytic, megakaryocytic, granulocytic (EMG) bone marrow proliferation described as panmyelosis by Dameshek [1,24]) was seen in 2 of 14 ET patients and in 11 of 16 PV and (Figure 1). Bone marrow histopathology appeared to be a powerful tool to differentiate ET and PV from all variants of primary or secondary erythrocytosis and reactive thrombocytosis with a sensitivity and specificity near to 100% [6,11,15,16,34]. The morphology of megakaryocytes were not different in ET and PV in the Rotterdam study (Figure 1, Tables 1-3). ET and PV patients in the Rotterdam ET/PV and the PVSG-01 PV studies show overlapping bone marrow pictures with increase of pleiomorphic clustered megakaryocytes (Figures 2,3).
Increased erythrocyte counts above 6x1012/L and increase of large pleiomorphic megakaryocytes in hypercellular bone marrow smears according to Dameshek (1950) [1,23] and bone marrow biopsy according to Michiels 1980 [2-3,34], appeared to a pathognomonic clue diagnostic for classical PV (Table 1). Increase of erythrocytes above 6x1012/L persisted during the iron deficient status in PV in remission by repeated venesection [23]. Two of our ET patients had borderline increased RCM, which was associated with erythrocytes above 6.0x1012/L, increased LAF score, and a negative bone marrow iron stain and should be diagnosed as PV. One ET case had erythrocyte count of 5.8x1012/L and increased LAF score developed PV within 2 years. Bone marrow iron stain was negative in all PV patients (N=16) and in 4 out of 12 evaluable ET patients and therefore cannot be used as diagnostic for PV. The most objective criterion to differentiate ET from PV (both may present typical ET/PV or PV. In recent studies we produced good evidence that the combination of bone marrow and erythrocyte count above 5.8X10/12L is diagnostic for PV (Table 3) [1,22].
European CLMP versus PVSG and WHO classification of the MPNs
Analysis of BM biopsies of 155 evaluated PV patients with documented increased RCM in the PVSG 01 study by Michiels using RCP and ECP criteria (Table 1) showed a spectrum of no, slight, moderate to marked (>80%) increase of bone marrow cellularity (Figures 2,3, Ellis 1986) [11], in ranges from <50-60% in 10, from 60-80% in 45 and from 80-100% in 100 cases (Figure 2). The silver stained reticulin fiber content was normal (RF 0 and 1) in 94, slightly increased (RF 2) in 40, and moderately to marked increased (RF 3 and 4) in 21 (Figure 2). Diagnosis according to bone marrow cellularity and morphology were typical for ET in 10, for ET/PV in 45, for PV in 70 and for PV/RF 3 or 4 in 13 PV patients when the RCP criteria were applied [11].
Between 2000 and 2015 we studied 10 JAK2V617F mutated patients with early stage ET or newly diagnosed PV who presented with migraine-like microvascular cerebral ischemic attacks (MIA) who were referred from various European countries to the Antwerp University Hospital for expert evaluation and treatment recommendation (Table 6). The PVSG diagnosis of 10 JAK2V617F mutated patients in the second international prospective clinical research study without use of bone marrow histopathology was ET in 6 and PV in 4 (Table 6). The 6 JAK2V617F mutated ET patients had erythrocyte counts below 6x1012/L. Three ET and 4 PV fulfilled the histological bone marrow features of typical PV according to ECMP bone marrow criteria [22,23]. Three ET patients with PV bone marrow features developed overt PV after long-term follow-up of 8, 9 and more than 10 years (slow onset PV, Figure 5, Table 6). Bone marrow cellularity was normal in 2 of 10 and increased (range 60-90%) due to increased erythropoiesis in 4 of 10 and increased erythropoiesis granulopoiesis in another 4 of 10 patients. Myelofibrosis (MF) MF-0 (RF 0/1) in 8 (5 ET, 3 PV), and MF-1 (RF 2) in MF-2 (1 ET, 1 PV) MPN patients. The diagnoses according to 2008 WHO criteria [24] were ET in 5, MNP unclassifiable in 1 and PV in 4. The diagnoses according to 2006-2015 ECMP criteria [34,46,47] (Table 6), were normocellular ET in 2, ET with features of early PV: low serum EPO, the presence of EEC and normal values for hemoglobin, hematocrit and erythrocyte counts (prodromal PV) in 3, hypercellular trilinear ET showing no leuko-erythroblastosis in 1, and acute onset PV not preceded by ET in 4 patients (Figure 5, Table 6).
| Table 6: European Clinical, molecular and pathological (ECMP) features of JAK2V617F mutated MPN in 6 ET patients and 4 patients with rapid onset PV (Personal observations by Michiels 2005-2012, at the Department of Hematology, Antwerp University Hospital, Antwerp, Belgium. | ||||||||||
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Age (years) and sex ( F/M) |
56/M | 60/M | 66/F | 47/F | 40/F | 31/F | 50/M | 43/F | 47/F | 38/M |
| Platelets at onset x109/ |
575 | 814 | 544 | 553 | 425 | 576 | 397 | 405 | 924 | 384 |
| JAK2V617F * | +/- | +/- | +/- | +/- | +/- | +/- | +/-à+/+ | +/- | +/+ | +/+ |
| Serum EPO | Normal | zero | decreased | decreasd. | nt. | decreased. | zero | decreased | zero | zero |
| Leukocytes x109/l | 6.7 | 5.3 | 12.9 | 8.2 | 6.1 | 6.2 | 7.3 | 14.3 | 13.1 | 8.0 |
| LAP score (N=<100) |
9nt | 160 | 197 | N | N | 186 | 163 | 263 | 232 | 284 |
| Hemoglobin g/dl | 13.6 | 15.5 | 14.2 | 14.4 | 13.4 | 14.0 | 18.6 | 17.3 | 16.3 | 12.8 |
| Hematocrit | 0.40 | 0.45 | 0.44 | 0.44 | 0.40 | 0.41 | 0.63 | 0.52 | 0.53 | 0.60 |
| Erythrocytes x1012/L | 4.5 | 5.3 | 4.7 | 4.8 | 4.6 | 4.9 | 6.3. | 6.1 | 7.4 | 6.7 |
| EEC | + | + | + | nt. | nt. | +. | + | + | + | + |
| Red cell mass | .N | N. | N | N | nt. | N. | ↑. | ↑ | ↑ | ↑ |
| Spleen, echogram cm |
. | 13 | 16 | 13 | 16.5 | 11.8 | 13.7 | 13 | 14.3 | 16 |
| BM diagnosis | ET | ET | PV | PV | EMGM | PV | PV | PV | PV | PV |
| Cellularity | 60% | 70% | 90% | 75% | 80% | 75% | 80% | 65% | 80% | 80% |
| M:E ratio | 1 | 1 | 1 | 0,5 | 4 | 0,7 | 0,7 | 1 | 1.5 | - |
| Myeloid lineage | N | N | ↑ | N | ↑ | N | N | N | ↑ | ↑ |
| Erythroid lineage | ↑ | ↑ | ↑ | ↑ | N | ↑ | ↑ | ↑ | ↑ | ↑ |
| Fibrosis | MF-0 | MF-1 | MF-0 | MF-0 | MF-0 | MF-0 | MF-0 | MF-0 | MF-1 | MF-0 |
| 2008 WHO diagnosis 2008 ECMP diagnosis Follow-up ECMP Diagnosis |
ET ET 1 4 yrs ET |
ET ET 1 12 yrs ET |
ET ET 2 10 yrs PV |
ET ET 2 11 yrs PV |
prePMF EMGM 15 yrs EMGM |
ET ET 2 8 yrs PV |
PV PV 4 yrs PV |
PV PV 1 yr PV |
PV PV 5 yrs PV |
PV PV 1 yr PV |
There are significant differences in blood and bone marrow features between ECMP defined ET compared to PVSG defined primary thrombocythemia hemorrhagica (PTH) at platelet counts above 1000x10/L (PTH) [8] described by Thiele (1988) in a cohort of 25 patients (Table in Figure 7) [25]. In PTH the values for hemoglobin, erythrocytes, leukocytes and LAP scores are normal [25], whereas LAP score were increased in RCP (Table 1) and ECMP defined ET [34,46,47] but in the normal range in PVSG defined PTH (Table in Figure 7). The megakaryocytes in PTH are large to giant with hyper-lobulated nuclei (Figure 7), whereas in RCP and ECMP defined JAK2V617F mutated ET patients the size of pleiomorphic megakaryocytes are medium similar to early and classical PV (Figure 4). In 2008 we studied bone marrow histopathology in 12 cases with JAK2 wild type ET carrying the MPL515 mutation (Table 4) kindly provided by the courtesy of Dr Vannucchi, Florence, Italy, Europe. Bone marrow pathology (P, Table 4) from patients with JAK2 wild type MPL515 mutated ET consistently displayed clusters small and large megakaryocytes with a greater number of giant megakaryocytes with hyper-lobulated stag-horn nuclei in a normal cellular bone marrow and no increase of erythropoiesis (Figure 8) [47].
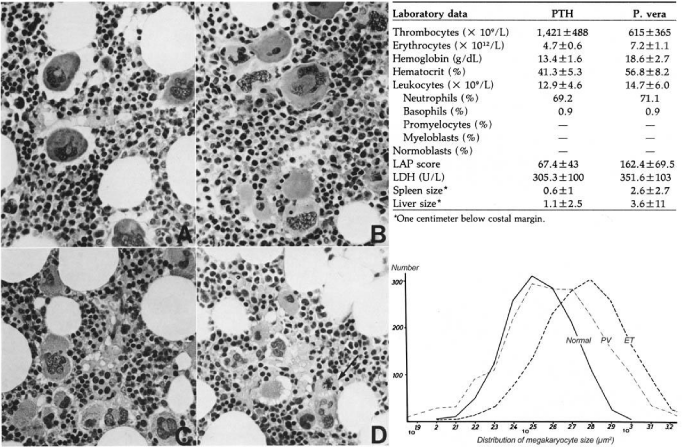
Figure 7: Megakaryocytic Leukemia (ML, Dameshek 1951) diagnosed as Primary Thrombocythemia Hemorrhagica (1975 PVSG) [8] compared to PV Thiele et al., demonstrated in 1988 [25] that in megakaryocytic leukemia (ML) [9] or PTH [8] the size of megakaryocytes in ML or PTH (upper panel) is larger than in PV (lower pannel) [25].
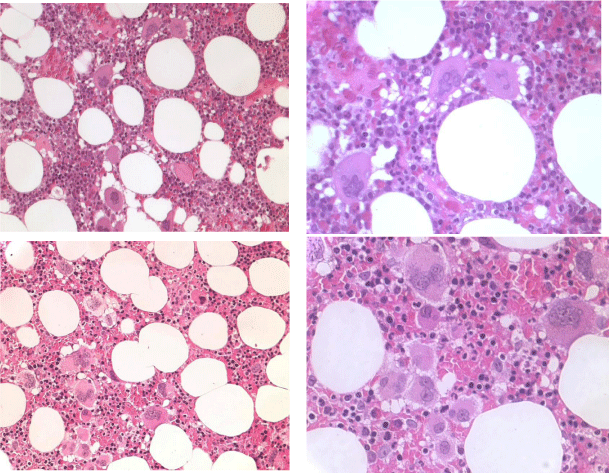
Figure 8: JAK2 wild type ET carrying the MPL515 mutation with enlarged and giant mature megakaryocytes with loose clusters of “staghorn-like” hyperlobulated nuclei. Case 1 and 2 Vannucchi, upper panels. Case 3, Vannucchi lower panels [48].
From 1994 to 2006, we observed a case of JAK2 wild type hemorrhagic thrombocythemia (HT) in a 9-year-old boy with a platelet count of 1596 to 1946x109/l, no splenomegaly on palpation, white blood differential count (metamyelocytes 0.5%, banded forms 1%, segmented granulocytes 52%, basophiles 2.5%, lymphocytes 35% monocytes 6%), low LAP score, and a hypercellular (80-100%) bone marrow with pre-fibrotic primary megakaryocytic and granulocytic myeloproliferative (PMGM) disease, absence of reticulin fibers, loose to dense clustering of enlarged dysmorphic megakaryocytes variable in size from large to giant with hyperploid and some naked nuclei (Figure 9). The dysmorphic megakaryocytes in this (Figure 9) and another case of PMGM (Figure 10) show definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear cytoplasmic ratio, which are not seen in MPL515 mutated normocellular ET (Figure 8) and also not in pre-fibrotic JAK2V617F mutated ET, ET/PV, PV and EMGM (masked PV) (Figure 5). Characteristic bone marrow findings of JAK2/MPL wild type, CALR mutated PMGM are consistent with pronounced thrombocythemia at platelet counts around 1000x109/ mimicking PTH as the presenting feature of pre-fibrotic PMGM [26,45,46], and do not meet the 2008 WHO criteria for AMM, or PMF [24]. The long-term follow-up of a typical case of PMGM showed normal blood cells counts, absence of the JAK2V617F mutation, without features of PV and myelofibrosis, and no splenomegaly on palpation (Figure 9). We observed similar cases of ECMP defined JAK2/MPL wild type CALR mutated PMGM (Figure 10), which in retrospect has been misinterpreted by Thiele and Vardiman as pre-fibrotic PMF [6,13,16] in the 2001 WHO classification. Tefferi and Thiele persisted to use the term pre-fibrotic and fibrotic stages of PMF without features of PVSG defined PV in the 2008 and 2016 WHO classifications [24,34].
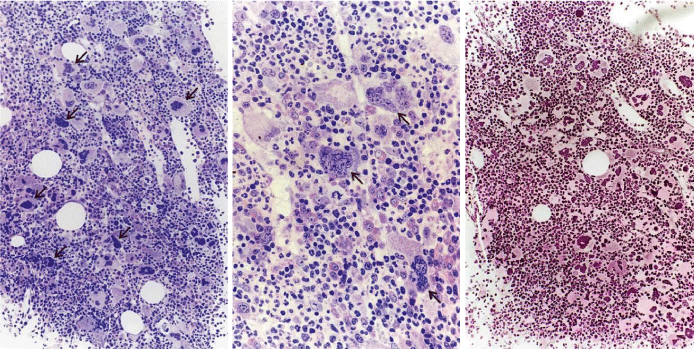
Figure 9: Third ET entity presenting with JAK2 wild type prefibrotic (right panel) primary megakaryocytic and granulocytic myeloproliferation (PMGM, left and middle), which is characterized by a hypercellular bone marrow due to dual myeloproliferation of granulopoiesis and dense clustered enlarged immature dysmorphic megakaryocytes (left and middle panels) with bulky (bulbous) hyperchromatic nuclei (arrows), which are never seen in JAK2 wild type MPL515 mutated ET and also not in the prefibrotic JAK2V617F mutated ET, prodromal PV, EMGM and trilinear PV entities.
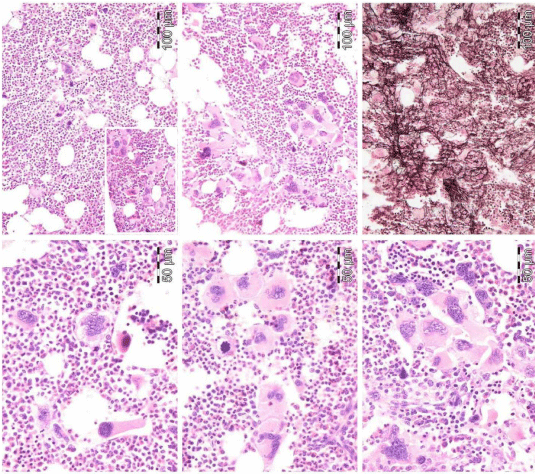
Figure 10: 37-years old woman (asymptomatic except fatigue) with JAK2 wild type thrombocythemia: platelets 1205 x10/9/L, Hb 12.5 g/dl, leukocytes 18 x10/9/L, slight increase of LDH, spleen not palpable but 13 cm on scan (normal value <12cm) as the presenting features of primary megakaryocytic granulocytic myeloproliferation (PMGM , dry tap; RF-2) diagnosed in 2013 as CALR mutated PMGM according to 2006-2018 ECMP criteria [34,47-50] and meeting the 2008/2016 WHO criteria for primary myelofibrosis (PMF) [24,29].
Discussion
Dameshek and Henthell (1940) [22] defined the diagnostic criteria for true polycythemia based on the description of 20 PV cases seen between 1928 and 1937, and proposed the historic set of symptoms, signs and laboratory tests for the diagnosis of PV1:

Dameshek (1940, 1950, 1951) demonstrated that the combination of plethoric appearance, splenomegaly, definitely elevated erythrocyte count in excess of 6x1012/L, elevated platelet count, elevated hematocrit is consistent with a definite diagnosis of PV1. The bone marrow is pathognomonic diagnositic for PV showing the combination of large megakaryocytes and panmyelosis (Dameshek) of increased trilinear erythrocytic, megakaryocytic and granulocytic (EMG) bone marrow proliferation without the need to measure blood volume [1,23]. According to Damehek in 1950 PV is a trilinear myeloproliferative disease (MPD) of erythrocythemia, thrombocythemia and granulocythemia in early pre-fibrotic stage of MPD disease, and complicated by spent phase PV and myeloid metaplasia of the spleen (increasing splenomegaly), and myelofibrosis in about one third of the cases after long-term follow-up of about 15 to 30 years [1,23].
The finding of increased LAF score and platelet count between 400 and 1000x109/ in the majority of our ET patients is a consistent finding in our ET and PV patients between 1975 to 2015 belong to the same JAK2V617F mutated MPN, whereas leukocyte count in ET usually normal or slightly increased. The combined use of bone marrow histology, blood cell (including erythrocyte) counts, EEC, serum EPO levels appeared to be highly sensitive and specific enough to distinguish the 3 stages of JAK2V617F mutated ET: normocellular ET, ET with features of PV (prodromal PV), and advanced ET with trilinear EMG bone marrow proliferation (Table 2). Interestingly, three cases of prodromal PV (ET with features of PV) indeed developed overt PV after long-term (around 10 years) follow-up (slow onset PV preceded by erythromelalgia or migraine-like atypical TIAs for more than 5 to10 years (Table 6). Four of 10 JAK2V617F mutated cases in our pilot study presented with acute onset PV not preceded by ET (Table 6).
The JAK2V617F mutated Thrombocythemia in trilinear MPN (Table 3), MPL515 mutated Thrombocythemia [34,46,47] and CALR Thrombocythemia associated with PMGM without features of PV clearly differ when the ECMP and 2018 CLMP criteria [51,52], are applied (Tables 4,5). The morphology of pleiomorphic megakaryocytes are not different between the JAK2V617F mutated MPNs in the three stages of normocellular ET, prodromal PV and masked PV (Figures 1,2,4,5, Table 3). The more the cellularity is increased, the more dysmorphic the megakaryocytes were in JAK2V617F mutated ET and PV. In JAK2V617F mutated hypercellular ET (EMGM or masked PV) pleiomorphic megakaryocytes show dysmorphic features of nuclei (not cloud-like) with a back ground of masked PV bone marrow histology [34,46,47]. Michiels et al., recognized that the MPL515 mutation Thrombocythemia is characterized by clusters of medium, large and giant megakaryocytes with a greater number of large deeply lobulated staghorn like nuclei, which are not seen in JAK2V617F mutated ET and PV patients [47]. Pich et al., described in 2012 characteristic bone marrow histopathology findings in 59 JAK2V617F positive ET and 44 JAK2 wild PT cases [27]. The bone marrow histology findings in the studies of Pich et al. [27], and Michiels et al. [34,46,47], in JAK2V617F mutated ET indeed have PV-like hypercellular morphological bone marrow changes showing pleiomorphic medium sized large mature megakaryocytes similar to our first 1980 cohort ET and PV (Figure 1) and also in PV patients of the PVSG 01 study (Figure 2) [11]. Clusters of large to giant megakaryocyte with staghorn nuclei and platelet count increase are more pronounced in MPL515 mutated normocellular Thrombocythemia with the complete absence of PV features [47]. ET with increased cellularity due to increased erythropoiesis is consistent with JAK2V617F mutated prodromal PV [34,46,47]. The prognosis of JAK2V617F mutated normocellular ET and prodromal PV is favorable and to be treated with low aspirin. Additional phlebotomy on top of low dose aspirin in early PV to maintain the hematocrit (Ht) around 0.40 in man and women is warranted for the cure and prevention of microvascular and major thrombosis [14,16]. The real field experiences from our prospective clinical observations in the 1980 and 1990s are completely in line with subsequent studies on patients with JAK2V617F mutated ET, prodromal PV and PV observed between 2005 and 2018 [27,34-46-50].
The 2008 and 2016 WHO-classifications of myeloproliferative neoplasms (MPN) are the direct extension of the 1975 PVSG classification and only based on clinical and laboratory features without the use of erythrocyte counts and bone marrow histology. The PVSG and WHO classification criteria are too crude to detect the early stages and of ET and PV and do not differentiate between normo-cellular ET, ET/PV (forme fruste PV) [39], classical PV and trilinear EMG (masked PV) bone marrow proliferation complicated by myelofibrosis [47-50]. The combined use of erythrocytes counts above 5.8x1012/L and bone marrow pathologic in the ECMP and CLMP classification is a pathognomonic clue to JAK2V617F mutated PV without the need of red cell mass (RCM) measurement. With the advent of JAK2V617F, MPL515 and CALR mutations as driver causes of three distinct MPN disease entities, ET is not essential and primary myelofibrosis (PMF) is not a primary disease anymore. MF is a secondary event in all molecular variants of MPD, MPN, CML and in hairy cell leukemia as well. Advanced MPN is featured by the combination of anemia, splenomegaly increased CD34 circulating cells, and myelofibrosis grade MF-2. MF-3 and MF-4 [6,16,17,24,29]. As 2006/2016 ECMP defined MF is a secondary event in advanced stages of all variant of MPN [26,34,50], this novel insight refers to the compelling need to better defined the natural history of JAK2V617F vs MPL515 vs CALR mutated MPN in large prospective clinical and basic research studies using the updated CLMP criteria including next generation sequencing and bone marrow pathology evaluation by expert pathologists. The JAK2V617F mutated ET, PV and MF appear to be a broad spectrum of sequential distinct trilinear MPN entities at the blood and bone marrow level during life-long follow-up [27,31-36,47-50]. By investigating the bone marrow features these JAK2V617F mutated cases in the initial pre-fibrotic stages some of them had a unique mono-linear megakaryocytic proliferation in a normocellular bone marrow lifelong in all three molecular variants of MPN. JAK2V617F mutated MPN had in addition a discrete expansion of the erythropoiesis in prodromal PV, and evolve into hypercellular trilinear megakaryocytic erythrocytic and granulocytic in classical, masked and advanced PV as can be documented by bone marrow proliferation and associated clinical features [32-37,47-50]. Increased erythrocytic bone marrow cellularity (60%-80%) in ET/PV patient was very well known to European pathologists [30,31]. Thiele et al., described the so-called initial (latent) stage of PV as ET mimicking PV [37]. The laboratory data of 23 cases diagnosed as initial or latent PV not meeting the 1975 PVSG and 2008 WHO criteria for PV did have increased erythrocyte count above 6x1012/L in men and above 5.5x1012/L in female, which indeed is consistent with the diagnosis of overt PV according to 1980 RCP and 2012-2018 ECMP criteria [37]. JAK2V617F mutated ET and prodromal PV are featured by increased LAF score, EEC+, low serum EPO and a normal erythrocyte count (<6x1012/L) [46,47].
The 2005 concept of Vainchenker & Constantinescu [33] is that heterozygous JAK2V617F mutation leading to constitutively activated megakaryocytes due to increased sensitivity to TPO is enough to induce ET phenotype [32,33,36]. The production of hyperreactive (sticky) platelet in thrombocythemia of ET and PV patients is the cause of platelet mediated arteriolar inflammation and thrombosis in ET and PV patients [34,35], (platelet thrombophilia Dames (1940) [1] or Sticky Platelet Syndrome, Michiels 1985) [2-4]. Thrombocythemia is associated with no or slight increased erythropoiesis in ET (ET stage 1, cases 1 and 2 of table 6), or definite increase of erythropoiesis in ET with features of PV (prodromal PV, cases 3 and 4 of table 6). Early stage erythromelalgic thrombotic JAK2V617F mutated ET with low JAK2V617F mutation load does exist [38] can now be diagnosed at as normocellular ET without progression into pro-dromal PV or classical PV during long-term follow-up of more than 20 years (Figure 5A). Homozygous JAK2V617F mutated hematopoietic progenitor cells are hypersensitive to EPO and TPO and do induce trilinear erythropoiesis, megakaryopoiesis and granulopoiesis (trilinear EMG) bone marrow proliferation. Such hypercellular trilinear EMG bone marrow pictures are the pathognomonic diagnostic clue for JAK2V617F mutated PV, masked PV or post-PV myelofibrosis without the need to measure red cell mass (RCM) [34-36,47-50]. The sequential occurrence of heterozygous/WT into homozygous JAK2V617F mutation due to mitotic recombination of chromosome 9p can readily explain the evolution of ET and prodromal PV into slow onset PV or masked PV with subsequent evolution into post-PV and post-ET myelofibrosis and splenomegaly due to myeloid metaplasia of the spleen (Table 6, Figure 6) [1,23,34,36,46-52]. Lifelong follow-up studies of slow onset PV and acute onset PV are lacking. There is a broad spectrum of transitional states between JAK2V617F mutated prodromal, masked and overt PV, its early reticulin fibrotic (RF) and reticulin collagen fibrotic (RCF) stages on one hand and post-ET and post-PV-myelofibrosis during long-term follow-up of 10 to even more than 25 years of follow-up were already defined by Dameshek (1950) [1,23]. The sequential use of RCP, ECP and 2006 ECMP and 2018 CLMP criteria did pick the early latent stages of all clonal variants and overt stages of MPN of various molecular etiologies [1,22,23,34,39-41,47,48].
We produced good evidence that ML (megakaryocytic leukemia) defined by Dameshek (1951) consist of MPL515 and CALR mutated Thrombocythemia without features of PV at diagnosis and during follow-up [42-44,48]. JAK2/MPL wild type cases of pre-fibrotic PMGM first defined by Georgii in 1990 [26] has recently been discovered by Michiels et al., as CALR mutated Thrombocythemia associated with PMGM [44,47,48]. Both MPL515 and CALR mutated thrombocythemia have no features of PV including low LAP score, whereas the incidence of secondary myelofibrosis is similar as in trilinear PV. In contrast myelofibrosis and palpable splenomegaly is rare in heterozygous JAK2V617F mutated ET [26,34,45-50] and much more prevalent in heterozygous/homozygous or homozygous JAK2V617F mutated hypercellular PV. The three main distinct MPNs at the level of bone marrow characteristics combined with MPN-specific clinical and laboratory features EEC, EPO levels in JAK2V617F mutated ET, prodromal PV and overt PV versus MPL515 and CALR mutated Thrombocythemia and Myelofibrosis without features of PV mutually exclude each other when the 2006/2018 ECMP [47-49] and 2018 CLMP [51,52], criteria are applied [47-52].
Acknowledgement
Contribution of authors
JJM, KL and FWJTK designed the study and JJM wrote the manuscript. JJM, ZB and WS collected the clinical data between 1975 and 2015. FWJTK evaluated the bone marrow biopsy specimens from the study in 1975-1985 and KL and HDR performed the bone marrow pathology studies between 2000 and 2018.
Dr. Juergen Thiele, Cologne, Germany as a most respected active member of the EWG.MPD (1998-2018) significantly contributed to the formulation of the ECP and ECMP criteria for the MPDs ET, PV, and PMGM or PMF and its differentiation from Ph+ ET, Ph+ CML, and the Ph-negative CMML, MDS and CNL just by bone marrow histopathology alone.
References
- Michiels JJ. Physiopathology, etiologic factors, diagnosis and course of polycythemia vera as related to therapy according to William Dameshek 1940-1950. Turkish J Hematol. 2013; 30: 102-110. Ref.: https://goo.gl/VWF3Td
- Michiels JJ, Ten Kate FWJ, Vuzevski VD, Abels J. Histopathology of erythromelalgia in thrombocythemia. Histopathology. 1984; 8: 669-678. Ref.: https://goo.gl/t79oZi
- Michiels JJ, Abels J, Steketee J, van Vliet HHDM, Vuzevski VD. Erythromelalgia caused by platelet mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med. 1985; 102: 466-471. Ref.: https://goo.gl/SctHNS
- Michiels JJ, Koudstaal PJJ, Mulder AH, Van Vliet HHDM. Transient neurologic and ocular manifestations in primary thrombocythemia. Neurology. 1993; 43: 1107-1110. Ref.: https://goo.gl/cYQy8p
- Michiels JJ, Juvonen E. Proposal for revised diagnostic criteria of essential thrombocythemia and polycythemia vera by the Thrombocythemia Vera Study Group (TVSG). Semin Thromb Hemostas. 1997; 23: 339-347. Ref.: https://goo.gl/9igXoi
- Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocthemia, polycythemia vera, and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol. 2002; 76: 133-145. Ref.: https://goo.gl/nZkVe3
- Michiels JJ, De Raeve H, Hebeda K, Lam KH, Berneman Z, et al. WHO bone marrow features and European clinical molecular and pathlogical (EMCP) criteria for the diagnosis and classification of myeloproliferative disorders. Leuk Res. 2007; 31: 1031-1038. Ref.: https://goo.gl/xNUgyu
- Laszlo J. Myeloproliferative disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD, and primary hemorrhagic thrombocythemia. Semin Haematol. 1975; 12: 409-432. Ref.: https://goo.gl/8pu88V
- Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951; 6: 372-375. Ref.: https://goo.gl/VVzyAJ
- Berlin MI. Diagnosis and classification of the polycythemias. Semin Hematol. 1975; 12: 339-351. Ref.: https://goo.gl/U5Yaad
- Ellis JT, Silver RT, Coleman M, Geller SA. The bone marrow in polycythemia vera. Semin Hematol. 1986; 12: 433-444. Ref.: https://goo.gl/fNNG28
- Kurnick JE, Ward HP, Block MH. Bone marrow sections in the differential diagnosis of polycythemia. Arch Pathol. 1972; 94: 489-499. Ref.: https://goo.gl/8vjxEJ
- Michiels JJ, Kutti J, Stark P, Bazzan M, Gugliotta L, et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thromboythemia, polycythemia vera and essential megakaryocytic granulocytic myeloproliferation and myelofibrosis. Neth J Med. 1999; 54: 46-62. Ref.: https://goo.gl/VbWuP2
- Michiels JJ, Barbui T, Finazzi G, Fruchtman SM, Kutti J, et al. Diagnosis and treatment of polycythemia vera and possible future study designs of the PVSG. Leukemia Lymphoma. 2000; 36: 239-253. Ref.: https://goo.gl/JA8arG
- Michiels JJ. Bone marrow histopathology and biological markers as specific clues to differential diagnosis of essential thrombocythemia, polycythemia vera and prefibrotic or fibrotic agnogenic myeloid metaplasia. The Hematol J. 2004; 5: 93-102. Ref.: https://goo.gl/C7nKXe
- Michiels JJ, Kvasnicka HM, Thiele J. Myeloproliferative Disorders. Current perspectives on diagnostic criteria, histopathology and treatment in essential thrombocythemia, polycythemia vera, and chronic idiopathic myelofibrosis. ISBN 3-9808075-6-8, 2005. Ref.: https://goo.gl/UvMStz
- Thiele J, Kvasnicka HM, Facchetti F, Franco V, Van Der Walt J, et al. European consensus for grading bone marrow fibrosis and assessment of cellularity in myeloproliferative diaorders. Haematologica. 2005; 90: 1128-1132. Ref.: https://goo.gl/AEWjiW
- Baxter EJ1, Scott LM, Campbell PJ, East C, Fourouclas N, et al. Acquired mutation of tyrosine kinase in human myeloproliferative disorders. Lancet. 2005; 365: 1054-1061. Ref.: https://goo.gl/JRaA1r
- Messinezy M, Westwood NB, Woodcock SP, Strong RM, Pearson TC. Low serum erythropoietin - a strong diagnostic criterion of primary polycythaemia even at normal haemoglobin levels. Clin Lab Haematol. 1995; 17: 217-220. Ref.: https://goo.gl/4QfJiu
- Messinezy M1, Westwood NB, El-Hemaidi I, Marsden JT, Sherwood RS, et al. Serum erythropoietine values in erythrocytoses and in primary thrombocythemia. Br J Haematol. 2002; 117: 47-53. Ref.: https://goo.gl/b3vQaJ
- Mossuz P1, Girodon F, Donnard M, Latger-Cannard V, Dobo I, et al. Diagnostic value of serum erythropoietine in patients with absolute erythrocytosis. Haematologica. 2004; 89: 1194-1198. Ref.: https://goo.gl/ezvfQq
- Dameshek W, Henstell HH. The diagnosis of polycythemia. Ann Intern Med. 1940: 13: 1360-1387. Ref.: https://goo.gl/9UY1MD
- Dameshek W. Physiopathology and course of polycythemia vera as related to therapy. JAMA. 1950; 142: 790-797. Ref.: https://goo.gl/sNRiJJ
- Thiele et al. 2008 WHO criteria for polycthemia vera, primary myelofibrosis and essential thrombocythemia. In: Swerdlow SH, Campo E, Harris NL, et al: WHO Classification of Tumours of Haematopoietic and Lympoid Tissues. Lyon France IARC Press. 2008; 40-50.
- Thiele J, Zankovich R, Schneider G, Kremer B, Fischer R, et al. Primary (essential) thrombocythemia versus polycythemia rubra vera. A histmorphometric analysis of bone marrow features in trephine biopsies. Anal Quant Cytol Histol. 1988; 10: 375-382. Ref.: https://goo.gl/MLursT
- Georgii A, Vykoupil KF, Buhr H, Choritz H, Doehler U, et al. Chronic myeloproliferative disorders in bone marrow biopsies. Path Res Pracxt. 1990; 186: 3-27. Ref.: https://goo.gl/mU4E5g
- Piche A, Riera L, Beggiato E, Nicolino B, Godio L, et al. JAK2V617F mutation and allele burden are associated with distinct clinical and morphological subtypes in patients with essential thrombocythemia. J Clin Pathol. 2012; 65: 953-954. Ref.: https://goo.gl/UrE18X
- Thiele, Kvasnicka HM. Chronic myeloproliferative disorders with thrombocythemia: a comparative study of two classification systems (PVSG, WHO) on 839 patients. Ann Hematol. 2003; 82: 148-152. Ref.: https://goo.gl/tv1drc
- Tefferi A1, Thiele J, Orazi A, Kvasnicka HM, Barbui T, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007; 110: 1092-1097. Ref.: https://goo.gl/HRpXUq
- Buhr T, Hebeda K, Kaloutsi V, Porwit A, Van der Walt J, et al. European Bone Marrow Working group trial to discriminate essential thrombocythemia from prefibrotic primary myelofibrosis. Haematologica. 2012; 97: 360-365. Ref.: https://goo.gl/3tXFk5
- Michiels JJ, Commandeur S, Hoogenboom GJ, Wegman JJ, Scholten L, et al. JAK2V617F positive early stage myeloproliferative disease (essential thrombocythemia) as the cause of portal vein thrombosis in two middle-aged women: therapeutic implications in view of the literature. Ann Hematol. 2007; 86: 793-800. Ref.: https://goo.gl/CCwbSU
- James C1, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, et al including Constantinescu S, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythemia var. Nature. 2005; 434: 1144-1148. Ref.: https://goo.gl/qHQJYj
- Vainchenker W, Constantinescu SN. A unique activating mutation in JAK2 (V617F) is at the origin of polycythemia vera and allows a new classification of myeloproliferative diseases. Hematology (Am Soc Hematol Educ Progr). 2005; 195-200. Ref.: https://goo.gl/5vUrwS
- Michiels JJ, De Raeve H, Berneman Z, Van Bockstaele D, Hebeda K, et al. The 2001 WHO and updated European Clinical and Pathological (ECP) criteria for the diagnosis, classification, and staging of the Philadelphia chromosome-negative chronic myeloproliferative disorders (CMPD). Sem Thromb Hemostas. 2006; 32: 307-340. Ref.: https://goo.gl/XNy2Qe
- Michiels JJ, Berneman Z, Van Bockstaele, Van Der Planken M, De Raeve H, et al. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications, and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Semin Thromb Hemostas. 2006; 32: 174-207. Ref.: https://goo.gl/8WHXgk
- Villeval JL, James C, Pisani D, Casadevall N, Vainchenker W. New insights into pathogenesis of JAK2V617F-positive myeloproliferative disorders and consequences for the management of patients. Sem Thromb Hemostas. 2006; 32: 341-351. Ref.: https://goo.gl/KzCMz2
- Thiele J, Kvasnicka HM, Diehl V. Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematol. 2005; 113: 213-219. Ref.: https://goo.gl/ZjSa5L
- Gale RE, Allen AJR, Nash MJ, Linch DC. Log-term serial analysis of X-chromosome inactivation patterns andJAK2 V617F mutant levels in patients with essential thrombocythemia show that minor mutant-positive clones can remain stable for many years. Blood. 2007; 109: 1241-1243. Ref.: https://goo.gl/wBiyPZ
- Campbell PJ1, Scott LM, Buck G, Wheatley K, East CL, et al. Definition of essential thrombocythemia and relation of essential thrombocythemia to polycythemia vera based on JAK2 V617F mutation: a prospective study. Lancet. 2005; 366: 1945-1953. Ref.: https://goo.gl/nfWb6b
- Vannucchi AM1, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, et al. Clinical profile of homozygous JAK2V617F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007; 110: 840-846. Ref.: https://goo.gl/SC6fAj
- Antonioli E1, Guglielmelli P, Poli G, Bogani C, Pancrazzi A,, et al. Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica. 2008; 93: 41-48. Ref.: https://goo.gl/5EXWA5
- Vannucchi AM1, Antonioli E, Guglielmelli P, Pancrazzi A, Guerini V, et al. Charateristics and clinical correlates of MPL515W>L/K mutation in essential thrombocythemia. Blood. 2008; 112: 844-847. Ref.: https://goo.gl/2bmSWK
- Beer PA1, Campbell PJ, Scott LM, Bench AJ, Erber WN, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008; 112: 141-149. Ref.: https://goo.gl/hVpnbo
- Klampf T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, et al. Somatic mutations od calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013; 369: 2379-2390. Ref.: https://goo.gl/eWxPi3
- Georgii A, Buhr H, Buesche G, Kreft A, Chorotz H. Classification and staging of Ph-negative chronic myeloproliferative diseases. Leuk Lymphoma. 1996; 22(suppl 1): 15-29. Ref.: https://goo.gl/vBXsjq
- Georgii A, Buesche G, Kreft A. The histopathology of chronic myeloproliferative diseases. Bailliere’s Clin Haematol. 1998; 11: 721-749. Ref.: https://goo.gl/a5Fzas
- Michiels JJ, Valster F, Wielenga J, Schelfout K, De Raeve H. European vs 2015 World Health Organization clinical molecular and pathological (WHO-CMP) classification of myeloproliferative neoplasms. World J Hematol. 2015; 4: 16-53. Ref.: https://goo.gl/iaCLC6
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H. Changing concepts of diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms: From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol. 2015; 133: 36-51. Ref.: https://goo.gl/CJRMmt
- Michiels JJ, Tevet M, Trifa A, Niculescu-Mizil E, Lupa A, et al. 2016 WHO Clinical Molecular and Pathological Criteria for Classification and Staging of Myeloproliferative Neoplasms (MPN) Caused by MPN Driver Mutations in the JAK2, MPL and CALR Genes in the Context of New 2016 WHO Classification: Prognostic and Therapeutic Implications. MAEDICA. 2016; 11: 5-25. Ref.: https://goo.gl/wJH2oJ
- Michiels JJ, De Rave H, Valster F, Potters V, Kim Y, et al. Extension of 2016 World Health Organization (WHO) classification and a new set of clinical, laboratory, molecular and pathological (CLMP) criteria for the diagnosis of myeloproliferative neoplasms: from Dameshek to Vainchenker, Green and Kralovics. EMJ. 2017a; 2: 72-81. Ref.: https://goo.gl/3QED5Z
- De Raeve H, Michiels JJ, Valster F, Potters V, Kim Y, et al. Novel clinical, laboratory, mollecular and pathological (2018 CLMP) criteria for the differential diagnosis of three distinct myeloprproliferative neoplasms: the role of driver mutation analysis and bone marrow histology. Int J Cancer Res Ther. 2018; 3: 1-13.
- De Raeve H, Fostier K, Valster F, Potters V, Kim Y, et al. Bone marrow histology is a pathognomonic clue to each of the JAK2V617F, MPL515 and CALR mutated thrombocythemia in myeloproliferative neoplasms. Clin Res Hematol. 2018; 1: 1-7. Ref.: https://goo.gl/hq1ZCN