Research Article
Primary myelofibrosis is not primary anymore since the discovery of MPL515 and CALR mutations as driver causes of mono-linear megakaryocytic and dual megakaryocytic granulocytic myeloproliferation and secondary myelofibrosis

Jan Jacques Michiels1* and Hendrik De Raeve2
1Goodheart Institute, Nature Medicine & Health, Hematology, Coagulation and Vascular Medicine Research Center, Freedom of Science and Education, European Free University Network, Rotterdam, Netherlands
2Department of Pathology, OLV Hospital Aalst and University Hospital Brussels, Belgium
*Address for Correspondence: Jan Jacques Michiels, Multidisciplinary Internist & Scientific Investigator, European Working Group on Myeloproliferative Neoplasm EWG.MPN 1998-2018, Goodheart Institute in Nature Medicine, Rotterdam, Europe, Erasmus Tower, Veenmos 13, 3069 AT Rotterdam, Netherlands; Tel: +31-626970534; Email: [email protected]
Dates: Submitted: 25 March 2019; Approved: 12 April 2019; Published: 15 April 2019
How to cite this article: Michiels JJ, De Raeve H. Primary myelofibrosis is not primary anymore since the discovery of MPL515 and CALR mutations as driver causes of mono-linear megakaryocytic and dual megakaryocytic granulocytic myeloproliferation and secondary myelopfibrosis. Int J Bone Marrow Res. 2019; 2: 018-026. DOI: 10.29328/journal.ijbmr.1001003
Copyright License: © 2019 Michiels JJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Abstract
Primary myelofibrosis (PMF) is a distinct clinicopathological myeloproliferatve disease (MPD) not preceded by any other MPD ET, PV, CML,... Combined use of bone marrow histology and increased erythrocyte counts above 5.8x1012/L can replace increased red cell mass at time of presentation as the pathognomonic clue for the correct diagnosis of hetero/homozygous or homozygous mutated PV. Erythrocyte counts are in the normal range below 5.8x1012/L in heterozygous JAK2V617F mutated ET and prodromal PV but above 5.8x1012/L in heterozygous-homozygous or homozygous mutated PV. The bone marrow cellularity and morphology in pre-fibrotic ET, prodromal PV and PV carrying the JAK2V617F mutation are overlapping showing clustered increase of large mature pleomorphic megakaryocytes (M) with no increase of cellularity (<60%) in ET. The bone marrow is hypercellular (60%-80%) due to increased erythropoiesis megakaryopoiesis (EM) in prodromal and classical PV and trilinear hypercellular (80%-100% due increased megakaryopoiesis, erythropoiesis and granulopoiesis (EMG) in advanced PV and masked PV. Bone marrow cellularity ranging from normal (<60%) in ET to increased erythropoiesis (EM) in prodromal PV to hypercellular (80-100%) in advanced PV and masked PV largely depends on increasing JAK2V617F mutation load from low to high on top of other biological MPN variables like constitutional symptoms during long-term follow-up. MPL515 mutated ET is featured by an increase of clustered small and giant megakaryocytes with hyper-lobulated staghorn-like nuclei in a normal cellular bone marrow. The third entity of pronounced JAK2/MPL wild type ET associated with primary megakaryocytic granulocytic myeloproliferation (PMGM) without PV features proved to be caused by calreticulin (CALR) mutation. CALR mutated thrombocythemia is characterized by dual proliferation of megakaryocytic and granulocytic bone marrow proliferation of dense clustered large to giant immature dysmorphic megakaryocytes with bulky (bulbous) hyperchromatic nuclei, which are not seen in MPL515-mutated Thrombocythemia and JAK2V617F-Thrombocythemia, prodromal PV and classical PV.
Introduction
Primary myelofibrosis (PMF) according to the criteria of the Polycythemia Vera Study Group (1975 PVSG) is a clinicopathological entity not preceded by any other MPD PV, CML, or MDS, and characterized by various degrees of anemia, splenomegaly, leuko-erythroblastosis, with tear drop-shaped erythrocytes, and dry tap on BM aspiration due to various degrees of myelofibrosis (MF) or osteosclerosis [1]. In 1977 Silverstein reviewed the characteristic findings in PMF [1,2]. All PMF patients, are usually above the age of 50 years, have large spleens, a leuko-erythroblastic blood reaction, striking teardrop poikilocytosis and dry tap on bone marrow aspiration. Under low-power magnification, a typical marrow section shows stranding, considerable fibrosis, and a few scattered megakaryocytes: at higher power, a dense fibrotic reaction is usually apparent. The marrow tends to be hypocellular in about 85%, normocellular in about 5% and hypercellular in about 10% of PMF patients. Anemia of ineffective erythropoiesis develops in about 60% of PMF patients within 5 to 10 years. Thrombocytopenia and leucopenia related to hypersplenism is seen in 30% and 14% of PMF patients. The diagnostic PVSG criteria for PMF have not been changed in the 2001, 2008 and 2016 WHO criteria for PMF [3-10].
Personal observations Rotterdam MPD study group 1975-2015
Case 1 in table 1 and related figure shows a typical case of primary pre-fibrotic PMF (prePMF) followed by progressive myelofibrosis, splenomegaly during 20 years follow-up. Sequential bone marrow aspirates and biopsies in 1971 and 1978 showed normal cellularity, no increase of erythropoiesis, selective increase of large megakaryocytes in a bone marrow smears and fine reticulin fibers in a bone marrow biopsy. In 1978, early stage PMF with reticulin fibrosis grade 2 was associated with significant splenomegaly but normal platelet counts and no anemia. Since 1985 full blown primary myelofibrosis (PMF) with progressive anemia and splenomegaly and leuko-erythroblastosis developed and the patient died in 1989 (Table 1). PMF was complicated by a transient episode of erythromelalgic thrombocythemia at platelet counts above 400x109/L at time of progressive PMF during the sequential fibrotic stages of increased reticulin fibrosis followed by collagen fibrosis. Pre-PMF with reticulin fibers grade 1 was diagnosed in 1971 in a 61-year-old man. The spleen had progressed to 7 cm in 1978 and to 13 cm below the costal margin in 1988. Since 1985 he suffered from aspirin responsive burning pain and red swelling of the right hand, fingers and left foot toes (erythromelalgia) at platelet counts of 400 to 500x109/L. After correction of increased platelet counts (800x109/L) by hydroxyurea to 200x109/L, erythromelalgia did not recur because anemia, splenomegaly and thrombocytopenia developed (Table 1).
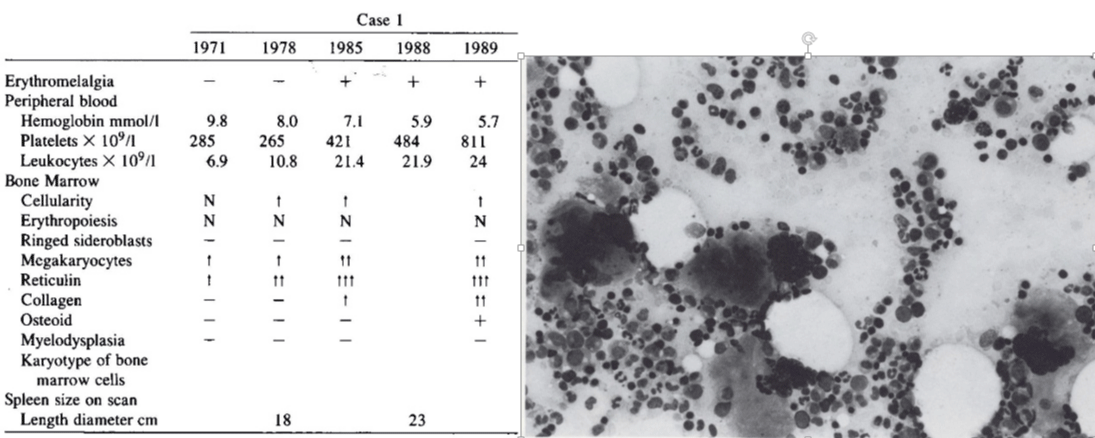
Table 1: Blood and bone marrow biopsy features in a case of pre-fibrotic and fibrotic stages of PMF during life-long follow-up and bone marrow smear at time of diagnosis in 1971. Source Michiels & Ten Kate Ann Hematol. 1992; 39: 131-136.
The PMF case in table 1 could be diagnosed as primary megakaryocytic granulocytic myeloproliferation (PMGM) when the Hannover Bone Marrow classification of the MPDs are applied [2]. Sequential bone marrow biopsies in 1985 and 1989 showed normal cellularity, coarse reticulin fibers, collagen fibrosis (with dry tap on aspiration), and increase of clustered large megakaryocytes. PMGM is misclassified as chronic idiopathic myelofibrosis (CIMF) by Thiele et al. in 1996, as CIMF in the 2001 WHO classification by Thiele & Vardiman and as PMF by Tefferi in the 2008 and 2016 WHO classifications. According to the 2006-2008 ECMP classification Michiels & De Raeve re-defined CIMF and PMF as JAK2 wild type PMGM (Table 2). PMGM has been recognized by Michiels in 2006 as JAK2/MPL wild type Thrombocythemia associated with PMGM since 2006 and as CALR mutated hypercellular thrombocythemia associated with PMGM since 2014 [11-18].
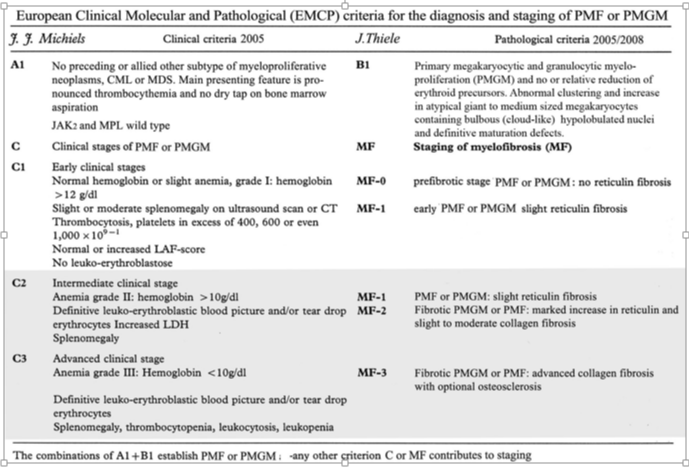
Table 2: European Clinical Molecular and Pathological (EMCP) criteria for the diagnosis and staging of PMF or PMGM.
JAK2V617F, MPL515 mutated and JAK2/MPL wild type MPN as the cause of PMF
MPL is the thrombopoeitin receptor (TPOR) on hematopoietic bone marrow cells discovered by William Vainchenker, who also discovered the JAK2V617F as the cause of trilinear myeloproliferative disorders: polycythemia vera (PV) and essential thrombocythemia (ET). About 40% of ET patients are JAK2 wild type. There is a small subgroup of 3% to 8.5% within JAK2 wild type ET patients, who carry an acquired gain of function MPL515 mutation as the cause of monolinear ET [8-10]. Tefferi from the division of Hematology, Mayo Clinic, Rochester, MN reviewed in January 2012, 1000 consecutive patients with primary myelofibrosis (PMF) seen at Mayo Clinic between November 4, 1977, and September 1, 2011 [3]. The International Prognostic Scoring System (IPSS), dynamic IPSS (DIPSS), and DIPSS-plus were applied for risk stratification. Separate analyses were performed for PMF patients seen at time of referral (N=1000), at initial diagnosis (N=340), and within or after 1 year of diagnosis (N=660). Anno 2012 there were 592 documented deaths and 68 leukemic transformations. Parameters at initial diagnosis vs time of referral included median age (66 vs 65 years), male sex (61% vs 62%), red cell transfusion need (24% vs 38%), hemoglobin level less than 10 g/dL (38% vs 54%), platelet count less than 100 × 109/L (18% vs 26%), leukocyte count more than 25 × 109/L (13% vs 16%), marked splenomegaly (21% vs 31%), constitutional symptoms (29% vs 34%), and abnormal karyotype (31% vs 41%). Mutational frequencies were 61% for JAK2V617F, 8% for MPL515, and 4% for IDH1/2. DIPSS-plus risk distributions at time of referral were low in 10%, intermediate-1 in 15%, intermediate-2 in 37%, and high in 37%. At that time CALR mutation as the driver cause of ET and PMF was not yet discovered. The corresponding median survivals were 17.5, 7.8, 3.6, and 1.8 years vs 20.0, 14.3, 5.3, and 1.7 years for patients younger than 60 years of age. Compared with both DIPSS and IPSS, DIPSS-plus showed better discrimination among high risk groups. Five-year leukemic transformation rates were 6% and 21% in low- and high-risk patients, respectively. Anno 2012 Dr. Tefferi concluded that prognosis in PMF should be assessed after a period of observation after diagnosis rather than at diagnosis. In addition, it seems that about 50% are treated in this manner whereas about 25% are candidates for allogenic stem cell transplant. Roughly 20% can be treated with a JAK2 inhibitor Jakafi/Ruxolitinib.
CALR mutations as the driver cause in JAK2/MPL wild type ET and PMF
The molecular etiology of JAK2/MPL wild type ET and PMF remained elusive untill Kralovics discovered the calreticulin (CALR) mutations as the driver cause in JAK2/MPL wild type ET and PMF patients negative for JAK2V617F and MPL515 mutation [4]. On behave of Austrian/Italian hematologists Dr Kralovics detected the CALR mutation in 78 of 311 (25%) ET patients and in 72 of 203 (35%) MF patients and in none of 382 PV patients. The CALR, JAK2V617F and MPL515 mutations mutually exclude each other since all ET and MF patients with CALR in exon 9 are JAK2 and MPL wild type [4,5]. Out of a large cohort of 289 JAK2/MPL wild type ET 195 (67%) carried one of the CALR mutations. Out of a cohort of 120 JAK2/MPL wild type MF a CALR mutation was detected in 105 (80%). Green and his team in the UK confirmed the high frequency of somatic CALR mutations in 70 to 84% of 120 ET or PMF patients negative for the JAK2/MPL mutation [6]. CALR mutations were present in 110 of 158 JAK2/MPL wild type MPN patients including 80 of 112 (70%) ET patients, 18 of 32 (56%) MF patients and in 12 of 13 patients with advanced PMF. CALR mutations were identified in 10 of 120 (8%) MDS patients (RA in 5 of 53, RARS in 3 of 27 and RAEB-T in 2 of 27), and in one patient each with chronic myelomonocytic leukemia (CMML) and atypical CML [6].
In a recent large study of Tefferi et al., 254 WHO-defined PMF patients carried the JAK2V617F-, MPL515- and CALR-mutation in 58%, 8.3% and 25% respectively and 8.7% were triple negative [7]. The median overall survival (OS) among 253 WHO-defined PMF patients in 83 CALR-, 21 MPL-, and 147 JAK2-mutated cases and in 22 triple negative cases was 8,2, 4.1, 4.3 and 2.5 years. As compared to CALR wild type MF, CALR-mutated MF patients were younger, had higher platelet count, lower leukocyte count, higher hemoglobin (less anemic) and lower DIPSS-plus score. Tefferi confirmed the findings of Kralovics that CALR-mutated MF patients had a favorable impact on median survival as compared to CALR-negative MF patients whether ASXL1-negative or positive. Among 181 WHO-defined CALR-negative MF patients, the median overall survival was 2.3 years in 55 CALR-negative/ASLX1-positive as compared to 5.6 years in 126 CALR-negative/ASXL1-negative MF patients. Among 146 cases of WHO-defined CALR-positive MF the median survival was 7 years in 20 CALR-positive/ASXL1-positive MF patients as compared to 9.6 years in 126 CALR-positive/ASXL1-negative MF patients.
By including bone marrow histology for ET, PV and PMF since 1980 Michiels & De Raeve changed the concept of PVSG-WHO defined Myeloproliferative Disorders (MPD) into the improved ECMP and WHO-CMP classifications of Myeloproliferative Neoplasms (MPN) [8-10]. The broad spectrum of JAK2V617F mutated trilinear MPN varied from ET, prodromal PV, masked PV, erythrocythemic PV, classical PV, and PV complicated by splenomegaly and myelofibrosis (MF). ET heterozygous for the JAK2V617F mutation is associated with low MPN disease burden and normal life expectancy. JAK2V617F mutation load increases from less than 50% in early stage PV to 100% in combined heterozygous/homozygous or homozygous JAK2V617F mutated advanced PV and post-PV MF. Pre-treatment bone marrow morphology and cellularity distinguish JAK2V617F mutated trilinear MPN from CALR and MPL mutated MPN. The morphology of clustered large pleomorphic megakaryocytes with hyper-lobulated nuclei are similar in JAK2V67F ET and PV patients (Figure 1). CALR mutated thrombocythemia shows bone marrow characteristics of PMGM (Table 2). CALR mutated thrombocythemia lacks features of PV and present with hypercellular thrombocythemia featured by dense clusters of immature large to giant megakaryocytes with hyperchromatic bulbous (cloudy) nuclei, which are characteristic for PMGM and not seen in JAK2V617F and MPL515 mutated MPN [8,9]. MPL515 mutated thrombocythemia typically shows mono-linear proliferation of large to giant megakaryocytes with hyper-lobulated staghorn like nuclei in a normocellular bone marrow without features of PV or PMGM. JAK2V617F mutation load in heterozygous mutated JAK2V617F normocellular ET ranges from low to 25%, in prodromal PV from 25% to 50% and increases to above 50% up to 100% in hypercellular heterozygous/homozygous or homozygous JAK2V617F mutated PV and advanced PV with myelofibrosis. The CALR and MPL515 mutation load is low in pre-fibrotic stages and rises from values below 25% to maximal values of around 50% in fibrotic stages of MPN [8,9]. JAK2V617F, CALR and MPL515 allele burden slowly increases together with the degree of splenomegaly, myelofibrosis and constitutional symptoms during life-long follow-up.
Bone marrow histology in MPL515 mutated normocellular ET
Bone marrow histology from a patient with JAK2 wild type ET carrying the MPLW515L mutation displayed clusters large megakaryocytes with a greater number of giant megakaryocytes with hyper-lobulated stag-horn nuclei in a normal cellular bone marrow and no increase of erythropoiesis (Figure 2) [10]. Michiels & De Raeve described the essential differences in bone marrow histopathology features of differential diagnostic significance between patients with MPL515 mutated (N=12) versus JAK2V617F mutated MPN (N=12) (Figure 2) [8-12]. The presence of clustered small and giant megakaryocytes with deeply lobulated stag-horn like nuclei (Figure 2) in ET carrying the MPL515 mutation are not seen in JAK2V617F positive prodromal PV, and classical PV (Figure 1). The pleomorphic medium to large megakaryocytes in JAK2V617F mutated ET and PV in bone marrow smears and bone marrow biopsy were similar in size and pleomorphy (Figure 1). There is local increase of erythropoiesis in areas of loose clustered pleiomorphic megakaryocytes in prodromal PV (Figure 1), which is not seen in MPL515 mutated ET (Figure 2). JAK2 wild type MPL515 mutated ET have no clinical, laboratory and bone marrow features of prodromal PV at diagnosis, do not evolve into PV during follow-up, and have normal LAP score, serum EPO and ferritin levels [1,5]. Laboratory and bone marrow histology evaluations have the diagnostic potential to separate the JAK2V617F mutated ET and prodromal PV with increased LAP score, low serum EPO and pleomorphic megakaryocyte morphology from MPL515 mutated ET with normal LAP score and serum EPO and giant mature megakaryocytes with staghorn-like nuclei [1,3,4]. The natural history of MPL515 patients clearly differ from JAK2V617F mutated ET and is typically featured by progressive anemia with increased reticuline fibrosis in a normocellular or may progress into refractory anemia in a hypocellular bone marrow and a high risk of MDS or leukemic transformation [10,11]. Pich et al., performed in 2012 a comparative bone marrow histology study in 103 WHO defined ET patients and demonstrated that the values for hemoglobin, hematocrit, and erythrocyte count, and bone marrow erythropoiesis were significantly lower in 44 JAK2V617F wild type ET as compared to 59 JAK2V617F mutated ET [13]. The megakaryocytes in JAK2V617F wild type patients were large to giant with more pronounced hyperlobulation of nuclei, whereas the megakaryocytes in JAK2V617F mutated ET and PV patients are medium sized to large (pleomorphic) with only a few giant forms [13].
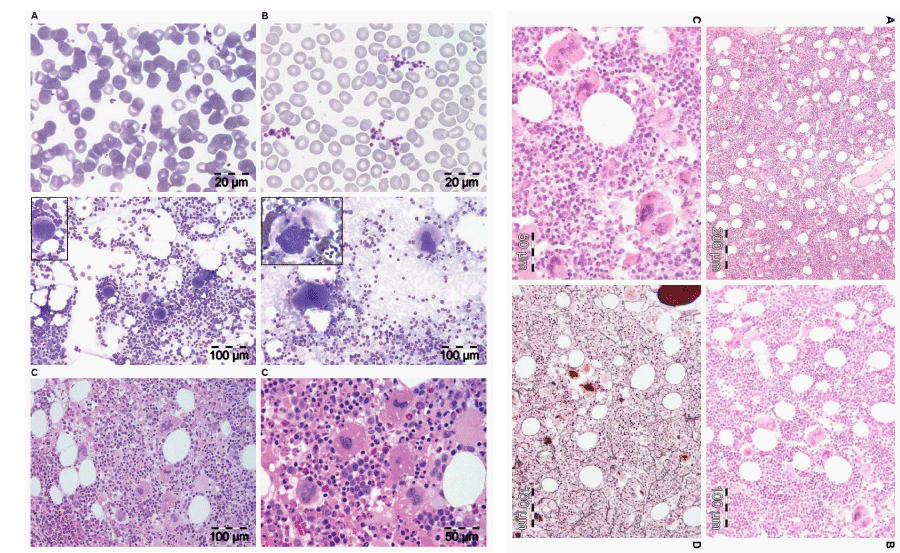
Figure 1: Left. Peripheral blood smears showing normal sized platelets in control (A) and large platelets in essential thrombocythemia (ET). Bone marrow smears (middle panels) showing normal sized megakaryocytes in control (left) and large megakaryocytes in ET (right). Bone marrow biopsy with hypercellular bone marrow histology (80% lower panels left ) due to increased erythropoietic and megakaryocytic (EM) proliferation showing increase of clustered large mature megakaryocytes with hyper-lobulated nuclei in JAK2V617F mutated ET with features of PV (low serum EPO, increased LAP score) consisted with prodromal PV. Right. Bone marrow biopsy showing a hypercellular bone marrow (90%) due to increased erythropoietic, megakaryocytic and granulocytic (EMG) proliferation with increase of clustered large mature megakaryocytes with hyper-lobulated nuclei and reticulin fibers grade 1 in JAK2V617F mutated classical PV and thrombocythemia with platelet of 1296x109/L.
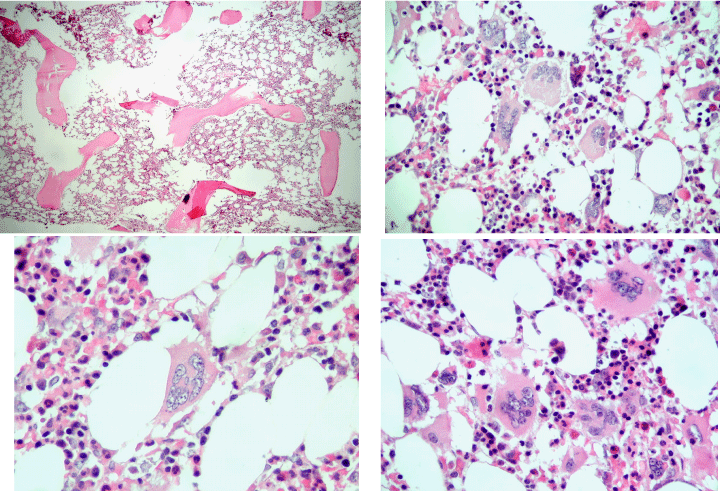
Figure 2: Bone marrow biopsy showing a normocellular bone marrow with no increase of erythropoiesis and increase of large to giant megakaryocytes with hyper-lobulated staghorn like nuclei in MPL515 mutated ET at platelet counts of 1385x109/L and no features of PV (normal serum EPO, LAP score and erythrocytes).
Bone marrow histology in CALR mutated JAK2 wild type ET and MF
Michiels & De Raeve studied between 2014 and 2018 twelve consecutive newly diagnosed CALR positive ET cases. We found a spectrum of two main stages of normocellular (40%-60%) ET and hypercellular bone marrow (60-80%) ET characteristics as the presenting feature of pre-fibrotic ET and hypercellular ET associated with a PMGM bone marrow picture (Table 2). Bone marrow histology in pre-fibrotic CALR normocellular ET (Figure 3) and in early fibrotic CALR myelofibrosis (MF, Figure 4) showed ‘dysmorphic’ large to giant megakaryocytes with definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear cytoplasmic ratio consistent with CALR mutated PMGM, which are not seen in MPL515 mutated ET and also not in JAK2V617F mutated ET, prodromal PV and classical PV. Pich performed in 2018 a comparative analysis of bone marrow cellularity, reticulin fibrosis, and the number, clustering and morphology, size of megakaryocytes and nuclei in 90 cases of WHO defined ET caused by the driver mutation JAK2V617F, CALR or MPL515 at time of diagnosis [14]. The JAK2V617F mutation was found in 58.9%, CALR in 28.9%, and MPL in 4.4% of the cases, and 7.8% were triple-negative. JAK2V617F mutated ET showed inceased bone marrow cellularity due to significant increase of erythropoiesis (Figure 3). MPL515 mutated ET showed the highest number of clustered large mature megakaryocytes with hyperlobulated staghorn-like nuclei in a normocellular or reduced cellular bone marrow (Figure 2). The bone marrow in CALR-mutated ET is normocellular or hypercellular due to dual megakaryocytic granulocytic myeloproliferation with no increase or reduced erythropoiesis CALR mutated ET in the Pich study showed dense clusters of large to giant dysmorphic megakaryocytes with hypo/hyperlobulated hyperchromatic bulbous or cloud-like nuclei similar as observed by Michiels & De Raeve (Figures 4-6) [10,11,15-18].
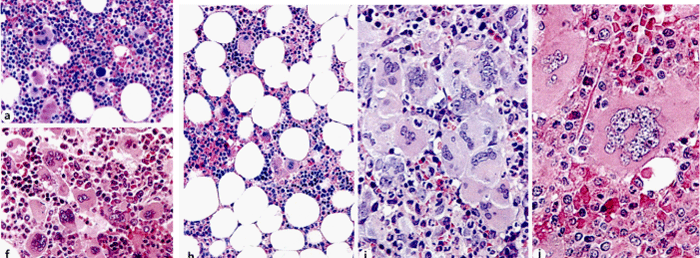
Figure 3: Bone marrow biopsy showing increased cellularity (70%) due to increased erythropoiesis and megakaryocytic (EM) proliferation with loos clusters of large megakaryocytes with hyper-lobulated nuclei in WHO defined JAK2V617F ET (a) in two studies of Piche 2008 and 2018. Dense clusters of large to giant megakaryocytes with hyperchromatic hypo/hyper-lobulated bulbous (cloudy) nuclei and relative reduction of erythropoiesis in WHO defined CALR mutated ET in the study of Pich 2018. Bone marrow biopsy with fields of normocellular and reduced cellularity (h) and the presence of dense clustered large to giant megakaryocytes with hyper-lobulated staghorn-like nuclei (i and j) in WHO defined MPL515 mutated ET in the study of Pich 2018.
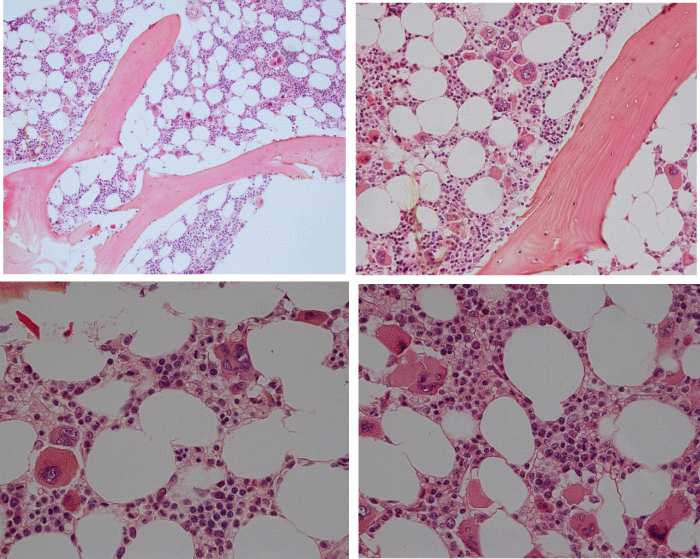
Figure 4: Bone marrow biopsy showing a normocellular bone marrow with loose and dense clusters of immature large to giant megakaryocytes with hypo/hyper-lobulated hyperchromatic cloudy (bulbous) nuclei in CALR mutated ET at platelet counts around 1000x109/L and no increase of reticulin fibers grade 0/1. Source Michiels & De Raeve.
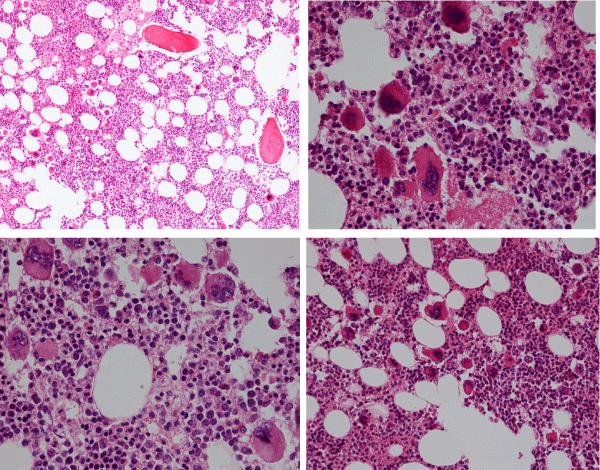
Figure 5: Bone marrow biopsy showing some increase of cellularity (60%) due to increase of primary megakaryocytic and granulocytic myeloproliferation (PMGM) with loose and dense clusters of immature large to giant megakaryocytes with hyperchromatic cloudy (bulbous) nuclei in CALR thrombocythemia (platelets 1296x10/L) and splenomegaly (spleen size 16 cm on echogram) complicated by acquired von Willebrand syndrome in the study o Michiels 2017.
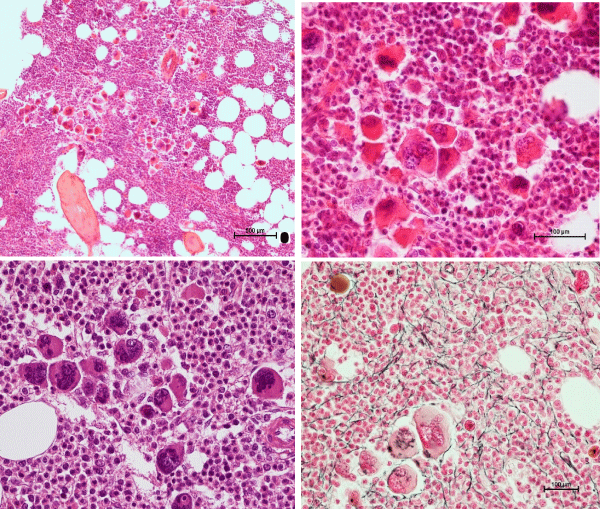
Figure 6: Bone marrow biopsy showing a hypercellular bone marrow (80%) due to primary megakaryocytic and granulocytic myeloproliferation (PMGM) with relative reduction of erythropoiesis and the presence of dense clustered immature megakaryocytes with hyperchromatic hypo/hyper-lobulated bulbous cloudy nuclei and slight increase og reticuline fibers grade 1 / 2 in CALR mutated thrombocythemia with moderate asymptomatic splenomegaly in the studies of Michiels & De Raeve 2018 [11,14-16].
Conclusion
With the advent of the MPL515, CALR and JAK2V617F mutations as the driver causes of three disctinct variant of ET (thrombocythemias) the term ET is not essential and PMF is not primary anymore simple because myelofibrosis is a secondary event in all three molecular variants of thrombocythemia and PV [15-18]. CALR-mutated hypercellular thrombocythemia and MPL515-mutated normocellular thrombocythemia are two distinct JAK2V617F wild MPN disease entities without features of PV and complicated by splenomegaly and secondary myelofibrosis during life-long follow-up. In conclusion, the term PMF is not primary anymore and should be replaced by CALR- thrombocythemia, MPL515-thrombocythemia and secondary MF similar as in JAK2V617F mutated post ET-MF and post-PV-MF as a secondary event [15-18].
References
- Silverstein MN. Myeloproliferative diseases. Hematology review. Postgraduate Medicine. 1977; 61: 206-201.
- Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol. 2002; 76: 133-145. Ref.: http://tinyurl.com/y3r2hp25
- Tefferi A1, Lasho TL, Jimma T, Finke CM, Gangat N, et al. One thousand patients with primary myelofibrosis: the Mayo Clinici experience. Mayo Clinic Proc. 2012; 87: 25-33. Ref.: http://tinyurl.com/y4eyy6nw
- Klampf Tl, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. New Eng J Med December. 2013; 369: 2379-2387. Ref.: http://tinyurl.com/y3hc7vte
- Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, et al. Jak2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcome. Blood, 2014; 123: 1552-1565. Ref.: http://tinyurl.com/y4vyyotn
- Nangalia J, Massie CE, Baxter J, Nice FL, Gundem G, et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Engl J Med. 2013; 369: 2391-2405. Ref.: http://tinyurl.com/yx8otrz7
- Tefferi A, Lasho TL, Finke CM, Knudson RA, Ketterling R, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014; 28: 1472-1477. Ref.: http://tinyurl.com/y6cpm8hn
- Michiels JJ, Hendrik De Raeve, Berneman Z, Van Bockstaele D, Hebeda K, et al. The 2001 world health organization and updated european clinical and pathological criteria for the diagnosis classification and staging of the Philadelphia-negative chronic myeloproliferative disorders. Sem Thromb Hemostas. 2006; 32: 307-340. Ref.: http://tinyurl.com/y2fnhwwo
- Michiels JJ, Forstier K, Valster F, Potters V, Schelfout K, et al. Criteria for the Classification and Staging of Five Distinct JAK2, MPL and CALR Mutated Myeloproliferative Neoplasms. J Hematol Thromb Dis. 2014; 2-6. Ref.: http://tinyurl.com/yy3mze9e
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H. Changing concepts and diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms: From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol. 2015; 133: 36-51. Ref.: http://tinyurl.com/y34d62ot
- Michiels JJ, Valster F, Wielenga J, Schelfout K, De Raeve H. European vs 2015 World Health Organization clinical molecular and pathological (WHO-CMP) classification of myeloproliferative neoplasms. World J Hematol. 2015; 6: 16-53. Ref.: http://tinyurl.com/y65e9q4h
- Michiels JJ, De Raeve H, Schwarz J, Campr V, Kim Y et al. Bone Marrow Histology Characteristics in MPL515 Mutated Thrombocythemia with Various Degrees of Myelofibrosis: A Cross Sectional Follow-up Study in Eight Cases. J Hematol Thrombo Dis. 2018; 6: 2. Ref.: http://tinyurl.com/yy8n4ygs
- Pich A, Riera L, Beggiato E, Nicolino B, Godio L, et al. JAK2V617F mutation and allele burden are associated with distinct clinical and morphological subtypes in patients with essential thrombocythemia. J Clin Pathol. 2012; 65: 953-954. Ref.: http://tinyurl.com/yxgbmga6
- Pich A, Riera L, di Celle PF, Beggiato E, Benevolo G, Godio L. JAK2V617F, CALR and MPL mutations and bone marrow histology in patients with essential thrombocythemia. Acta Haematol. 2018; 140: 234-239. Ref.: http://tinyurl.com/y5faxo7k
- Michiels JJ, Valster F, Potter V, Schelfout K, Schroyens W, et al. Secondary myelofibrosis in the natural history of JAK2, MPL and CALR mutated myeloproliferative neoplasms. J Hematol Thrmb Dis. 2016; 4: 3. Ref.: http://tinyurl.com/yxdsyxkj
- De Raeve H, Fostier K, Valster F, Potters V, Kim Y, et al. Bone Marrow Histology is a pathognomonic clue to each of the JAK2V617F, MPL,515 and Calreticulin Mutated Thrombocythemia in Myeloproliferative Neoplasms. Clin Res Hematol. 2018; 1: 1-7. Ref.: http://tinyurl.com/yycbcvnn
- De Raeve H, Michiels JJ, Valster F, Potters V, Kim Y, et al. Novel Clinical, Laboratory, Molecular and Pathological (2018 CLMP) Criteria for the Differential Diagnosis of three Distinct JAK2, CALR and MPL MutatedMyeloproliferative Neoplasms: The Role of Driver Mutation Analysis and Bone Marrow Histology. Int J Cancer Res Ther. 2018; 3: 1-12.
- Michiels JJ, Berneman Z, Schroyens W, J ten Kate FW, et al. European Clinical Laboratory, Molecular and Pathological (ECMP) criteria for prefibrotic JAK2V617F-Thrombocythemia and Polycythemia Vera versus MPL515 and CALR mutated Thrombocythemia and Myelofibrosis: From Dameshek to Michiels 1950-2018. Int J Bone Marrow Res. 2019; 2: 001-017. Ref.: http://tinyurl.com/yxvu6d8p