Research Article
The PVSG/WHO versus the Rotterdam European clinical, molecular and pathological diagnostic criteria for the classification of myeloproliferative disorders and myeloproliferative neoplasms (MPD/MPN): From Dameshek to Georgii, Vainchenker and Michiels 1950-2018

Jan Jacques Michiels1,4,5* and Hendrik De Raeve2,3
1Goodheart Institute, Nature Medicine & Health, Hematology, Coagulation and Vascular Medicine Research Center, Freedom of Science and Education, European Free University Network, Rotterdam, Netherlands
2Department of Pathology, OLV Hospital Aalst and University Hospital Brussels, Belgium
3European Working Group on Myeloproliferative Disorders (EWG.MPD), Belgium
4International Collaborations and Research on Myeloprolferative Neoplasm, Netherlands
5ICAR.MPN, Netherlands
*Address for Correspondence: Jan Jacques Michiels, MD, PhD Multidisciplinary Internist, Goodheart Institute in Nature Medicine & Health, Erasmus Tower, Veenmos 13, 3069 at Rotterdam, Netherlands, Tel: +31-626970534; Email: [email protected]
Dates: Submitted: 27 March 2019; Approved: 15 April 2019; Published: 17 April 2019
How to cite this article: Michiels JJ, De Raeve H. The PVSG/WHO versus the Rotterdam European clinical, molecular and pathological diagnostic criteria for the classification of myeloproliferative disorders and myeloproliferative neoplasms (MPD/MPN): From Dameshek to Georgii, Vainchenker and Michiels 1950-2018. Int J Bone Marrow Res. 2019; 2: 027-050. DOI: 10.29328/journal.ijbmr.1001004
Copyright License: © 2019 Michiels JJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Myeloproliferative disorders; Essential thrombocythemia; Polycythemia vera; Chronic idiopathic myelofibrosis; JAK2 V617F mutation; Bone marrow pathology

Abstract
The present article extends the PVSG-WHO criteria into a simplified set of Rotterdam and European Clinical, Molecular and Pathological (RCP/ECMP) criteria to diagnose and classify the myeloproliferative neoplasms (MPNs). The crude WHO criteria still miss the masked and early stages of ET and PV. Bone marrow histology has a near to 100% sensitivity and specificity to distinguish thrombocythemia in BCR/ABL positive CML and ET, and the myelodysplastic syndromes in RARS-T and 5q-minus syndrome from BCR/ABL negative thrombocythemias in myeloproliferative disorders (MPD). The presence of JAK2V617F mutation with increased erythrocytes above 6x1012/L and hematocrit (>0.51 males and >0.48 females) is diagnostic for PV obviating the need of red cell mass measurement. About half of WHO defined ET and PMF and 95% of PV patients are JAK2V617F positive. The combination of molecular marker screening JAK2V617F, JAK2 exon 12, MPL515 and CALR mutations and bone marrow pathology is 100% sensitive and specific for the diagnosis of latent, early and classical ECMP defined MPNs. The translation of WHO defined ET, PV and PMF into ECMP criteria have include the platelet count above 350 x109/l, mutation screening and bone marrow histology as inclusion criteria for thrombocythemia in various MPNs. According to ECMP criteria, ET comprises three distinct phenotypes of true ET, ET with features of early (“forme fruste” PV), and ET with a hypercellular erythrocythemic, megakaryocytic granulocytic myeloproliferation (EMGM or masked PV). The ECMP criteria clearly differentiate early erythrocythemic, prodromal and classical PV from congenital polycythemia and idiopathic or secondary erythrocytosis. The burden of JAK2V617F mutation in heterozygous ET and in homozygous PV is of major clinical and prognostic significance. JAK2 wild type MPL515 mutated normocellular ET and MF lack PV features in blood and bone marrow. JAK2/MPL wild type hypercellular ET associated with primary megakaryocytic granulocytic myeloproliferation (PMGM) is the third distinct CALR mutated MPN. The translation of WHO into ECMP criteria for the classification of MPNs have a major impact on prognosis assessment and best choice for first line non-leukemogenic approach to postpone potential leukemogenic myelopsuppressive agents as long as possible in ET, PV and PMGM patients.
Introduction
In the 19th century chronic myeloid leukemia (CML) and polycythemia vera (PV) have been described as primary distinct disease entities [1-3]. In 1960 Nowell and Hungerford described the presence of a minute chromosome in leukemic cells of patients with CML [4]. This minute chromosome was called Philadelphia (Ph) chromosome after the city of discovery [4]. Using banding techniques Janet Rowley (1973) discovered that the Ph chromosome originated from a translocation between the long arms of chromosomes 9 and 22, t(9;22)(q34;q11) [5]. Two groups collaborating in scientific friendship discovered that a hybrid gene is generated by the translocation consisting of the BCR gene on chromosome 22 and the ABL oncogene originating from chromosome 9 [6]. This results in a BCR/ABL fusion gene with high tyrosine kinase activity and CML-transformation capacity [7,8]. Ninety-five percent of all CML patients are Ph+; 90% are Ph+/BCR/ABL+, 5% are Ph-/BCR/ABL+, and 5% are Ph-/BCR/ABL-, the latter group usually diagnosed as atypical CML, juvenile CML, chronic neutrophilic leukemia or chronic myelomonocytic leukemia [9]. According to strict morphological, biochemical, cytogenetic and molecular criteria including the Ph+ chromosome and BCR/ABL fusion gene and protein, CML is a malignant disease with an obligate transition into acute leukemia, whereas PVSG defined essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (PMF) form the Ph-chromosome and BRC/ABL negative chronic myeloproliferative disorder (MPD) featured by a benign proliferation of the three hematopoietic cell lines [10].
In 1950, Dameshek defined PV as a chronic disorder of the bone marrow characterized by excessive production of nucleated red cells, granulocytes and megakaryocytes, peripheral blood erythrocytosis, leukocytosis and thrombocytosis [11]. Some cases however show pronounced elevation of erythrocytes or extreme degree of thrombocytosis, while in others the leukocyte counts may be at or close to leukemic levels, with only slight increase in red cells or platelets [11]. Regarding etiology of PV, Dameshek proposed in 1950 two highly speculative possibilities: first, the presence of excessive bone marrow stimulation by an unknown factor or factors, and second, a lack or a diminution in the normal inhibitory factor or factors (Figure 1) [11]. The discovery of the JAK2V617F mutation in 2005 by Vainchenker confirmed the hypothesis of Dameshek by demonstrating that the JAK2V617F mutation induced loss of inhibitory activity of the JH2 pseudokinase part on the JH1 kinase part of JAK2, leading to enhanced activity of the normal JH1 kinase activity of JAK2 (Figure 1) [12,13]. This renders the receptors of mutated hematopoietic stem cells hypersensitive to hematopoietic growth factors thrombopoietin (TPO), erythropoietin (EPO) and granulocyte colony stimulating factor (GCSF), resulting in trilinear myeloproliferation [12,13].
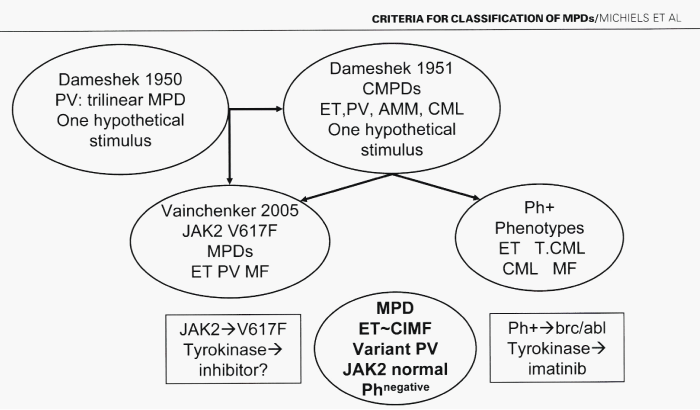
Figure 1: Left. Conceptual change of myeloproliferative disorders (MPD) into myeloproliferative neoplasms: From Dameshek 1950 to Vainchenker 2005 and Michiels 2008. According to Dameshek in 1950 polycythemia vera (PV) is a trilinear MPD due to one hypothetical stimulus has been confirmed by Vainchenker in 2005 to be caused by the JAK2V617F mutation as the cause of trilinear MPNs essential thrombocythemia (ET), PV and secondary myelofibrosis (Michiels 2006). The speculation of Dameshek in 1951 on the MPDs chronic myeloid leykemia (CML), ET, PV and agnogenic myeloid metaplasia (AMM) has been changed into Ph-postive ET and CML vs Ph-negative ET, PV and AMM by the discovery of t(9:22)(q34:q11) in Ph positive CML by Rowley and the BCR/ABL fusion gene and protein, a constitutively activated tyrokinase activity as the cause of Ph-positive CML and ET in the 1980s by Heisterkamp, Groffen & Grosveld Erasmus University Rotterdam in collaboration with Abels, Hagemeyer & Michiels from the Hematology Department Academic Hospital, Dijkzigt, Rotterdam, Netherlands.
The morphological distinction between Ph+ and BCR/ABL+ ET and thrombocythemia associated with Ph+ and BCR/ABL+ CML versus the Ph-negative thrombocythemia in various MPDs is primarily based upon conspicuous differences in the form and size of megakaryocytes in bone marrow smears and sections of bone marrow biopsy [10]. This difference observed by Michiels et al. in 1987, is obvious and reproducible in bone marrow biopsies, which enables pathologists to distinguish between small megakaryocytes in Ph+ disease versus large megakaryocytes with more or less pronounced hyper-lobulated nuclei in Ph-negative MPDs ET and PV (Figure 2) [10]. This original observation by Michiels (1987) and Georgii (1990) of diagnostic differentiation between Ph+ CML and Ph-negative MPD and subclassification of the MPDs into ET, classic PV and prefibrotic agnogenic myeloid metaplasia (AMM) or chronic idiopathic myelofibrosis (CIMF) was worked out by Thiele et al. in 1988 and 1989, (Figure 2) [14-17]. The criteria of the Polycythemia Vera Study Group (PVSG) and the 2001 WHO criteria to classify the MPDs as ET, PV and CIMF are suboptimal and overlook the early prefibrotic stages (Figure 2) [19-23]. Georgii et al. in 1990 [18], changed the PVSG-WHO defined ET, PV and PMF into the Hannover Bone Marrow Classification ET, PV and chronic megakaryocytic granulocytic myeloproliferation (PMGM) as three disctinct MPDs. In the present manuscript we propose to integrate the PVSG-WHO clinical and Hannover bone marrow criteria into the Rotterdam and European Clinical, Molecular and Pathological (ECMP) classification by including bone marrow pathology and the use of new laboratory and molecular markers for diagnostic differentiation of each of the latent (masked), early and overt MPDs.
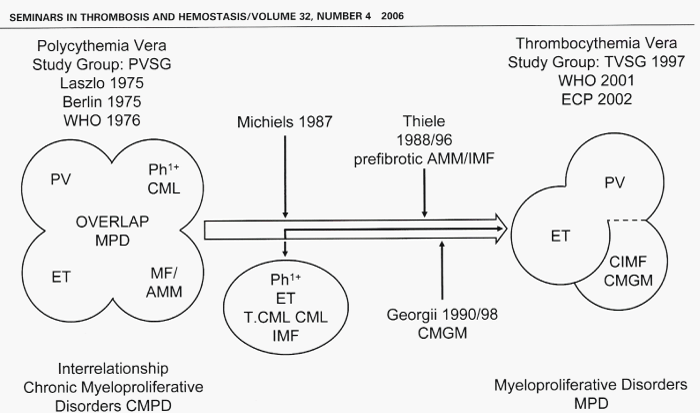
Figure 2: The concept of Polycythemia Vera Study Goup (PVSG) on the overlap myeloproliferative disorders (MPD) essential thrombocythemia (ET), polycythemia vera (PV), agnogenic myeloid metaplasia (AMM) and chronic myeloid leukemia (CML) was based on the 1951 Dameshek concept that the overlap MPD may be due to one hypothetical stimulans, which in retrospect appeared to be incorrect. The unifying concept of Dameshek in 1951 on the chronic myeloproliferative disorders (MPDs) PV ET AMM and CML has been broken up by the 1975 PVSG criteria by Michiels in 1987 and Georgii in 1990 into Ph-positive essential thrombocythemia (ET) and CML complicated by myelofibrosis (MF) and the Ph-negative MPDs ET, PV and MF either positive or negative for the acquired JAK2V617F mutation.
PVSG, TVSG, RCP and WHO criteria for the diagnoses of ET, PV and AMM
In 1986, the PVSG reduced the platelet count from 1000 to 600 x109/l as the arbitrary minimum for the diagnosis of ET [22,23]. In Since 1997 we defined the clinical and pathological characteristics of the Ph-negative MPDs by including bone marrow histopathology according to Georgii et al on top of PVSG criteria for ET, PV and CMGM [24]. The TVSG and RCP criteria combined a typical ET histological bone marrow picture with platelet counts in excess of 600 and 400 x109/l respectively (Table 1) [25,26]. The PVSG-WHO criteria comprises three phenotypes of prefibrotic early MPD: ET, early thrombocythemic PV, PV and prefibrotic and early fibrotic stages of PMF MF-0 and MF-1 [27-29]. Lengfelder et al., demonstrated that the minimum of 600x109/l platelets according to the 1986 PVSG criteria excluded early (masked) ET at platelet count below 600 x109/l in 29% of 143 ET cases [30]. In this study, 97% of all 143 ET patients showed a typical bone marrow histology of increase and clustering of enlarged megakaryocytes diagnostic for MPD [30]. This was associated with normal cellularity in 52% consistent with true ET, with increased erythropoiesis in 17% consistent with early PV, and with increased cellularity due to pronounced granulopoiesis in 45% consistent with pre-fibrotic PMF [30]. The 1986 PVSG-2001WHO criteria overlook latent and early stages of MPD in patients with thrombocythemia: First, initial ET with a typical ET bone marrow but platelet count below 600 x109/l; Second, initial PV with a typical PV bone marrow, platelet count less than 600 x109/l, low serum erythropoietin (EPO), normal red cell mass and hematocrit less than 0.51; Third, initial masked MPD with splenomegaly and normal or slightly increased platelet count and haematocrit [25]. The 2008 WHO extended the 1975 PVSG and 2001 WHO classification and changed the term MPD into myeloproliferative neoplasia (MPN) for the clinical diagnosis of ET, PV, PMF [29-33].

Table 1: The 1980-2005 Rotterdam Clinical and Pathological (RCP) criteria for Essential Thrombocythemia: ET. The 1980-2005 Rotterdam Clinical and Pathological (RCP) criteria for Polycythemia Vera: PV
In 1986, the PVSG reduced the platelet count from 1000 to 600 x109/l as the arbitrary minimum for the diagnosis of ET [22,23]. In Since 1997 we defined the clinical and pathological characteristics of the Ph-negative MPDs by including bone marrow histopathology according to Georgii et al on top of PVSG criteria for ET, PV and CMGM [24]. The TVSG and RCP criteria combined a typical ET histological bone marrow picture with platelet counts in excess of 600 and 400 x109/l respectively (Table 1) [25,26]. The PVSG-WHO criteria comprises three phenotypes of prefibrotic early MPD: ET, early thrombocythemic PV, PV and prefibrotic and early fibrotic stages of PMF MF-0 and MF-1 [27-29]. Lengfelder et al., demonstrated that the minimum of 600x109/l platelets according to the 1986 PVSG criteria excluded early (masked) ET at platelet count below 600 x109/l in 29% of 143 ET cases [30]. In this study, 97% of all 143 ET patients showed a typical bone marrow histology of increase and clustering of enlarged megakaryocytes diagnostic for MPD [30]. This was associated with normal cellularity in 52% consistent with true ET, with increased erythropoiesis in 17% consistent with early PV, and with increased cellularity due to pronounced granulopoiesis in 45% consistent with pre-fibrotic PMF [30]. The 1986 PVSG-2001WHO criteria overlook latent and early stages of MPD in patients with thrombocythemia: First, initial ET with a typical ET bone marrow but platelet count below 600 x109/l; Second, initial PV with a typical PV bone marrow, platelet count less than 600 x109/l, low serum erythropoietin (EPO), normal red cell mass and hematocrit less than 0.51; Third, initial masked MPD with splenomegaly and normal or slightly increased platelet count and haematocrit [25]. The 2008 WHO extended the 1975 PVSG and 2001 WHO classification and changed the term MPD into myeloproliferative neoplasia (MPN) for the clinical diagnosis of ET, PV, PMF [29-33].
The 1975 PVSG criteria followed the recommendations of Dameshek to define PV by increased red cell mass (erythrocytosis or erythrocythemia) and did not include bone marrow histology as a clue to MPD [11,34-36]. The PVSG investigators Wasserman and Berlin introduced 3 major and 4 minor clinical criteria as inclusion criteria the diagnosis of PV in the PVSG-01 study of which increased RCM was mandatory [19,20]. Increased RCM in PV patients corresponded to hematocrit values between 0.48 and 0.76 in all, platelet count above 400 x109/L in two-third and palpable spleen in two-third of about 400 PV patients in the PVSG-01 study [20]. One third of PV patients had normal platelet count and spleen size at time of presentation. Pure erythrocythemia featured by increased hemoglobin, hematocrit and red cell mass but normal leukocytes, thrombocytes and spleen size was labelled as idiopathic erythrocytosis (IE) [37] or pure erythrocythosis [38]. Minor B criteria did appear in untreated IE patients during follow up and was associated with a high incidence of major or lethal cerebrovascular thrombotic disease [37]. This category of IE or the earliest erythrocythemic stage of PV comprises about 10 to 20% of the PV cases at time of presentation was completely overlooked by the PVSG criteria [37,38]. In the 1970s tools available in the USA to differentiate idiopathic erythrocytosis into myeloproliferative PV versus primary or secondary erythrocytosis include pretreatment bone marrow biopsy [39-41] and EEC [42-45]. Such a powerful diagnostic differentiation has been implemented in European Clinical and Laboratory (ECP) criteria of the MPD ET, PV and myelofibrosis (MF) since the 1990s [24-26]. Clinicians and pathologists should be aware that pre-treatment bone marrow biopsy specimens in 191 PV patients of the PVSG-01 study [40,41] with increased RCM showed a normal bone marrow cellularity with no increase of normal or clustered large megakaryocytes (idiopathic erythrocytosis) in about 7.5%, slight to moderate increased bone marrow cellularity (60-80%) and increase of clustered large megakaryocytes in two-thirds (consistent with early erythrocythemic, thrombocythemic stage PV), and pronounced trilineage hypercellularity (80-100%) of the bone marrow in one-third consistent with classic PV with trilinear “panmyelosis” as described by the PVSG [35,40,41] exactly as defined at the bone marrow level by Georgii et al. [18], Michiels & Thiele [46,47] and by Thiele et al. [46-50].
The 1980-2005 Rotterdam Clinical and Pathological (RCP) criteria revealed that various degrees of characteristic PV bone marrow histology features (irrespective of RCM measurements) are seen in ET and three different PV stages of newly diagnosed MPD patients (Table1) [32,33,36,46,47]. First, early thrombocythemic prodromal PV mimicking ET with a hematocrit in the upper limit of normal (<0.50) and erythrocytes below 6x1012/L but increased platelet count (>400 x109/l) without or with slight splenomegaly (prodromal PV). Second, “idiopathic erythrocytosis” with increased RCM, high hematocrit, low serum EPO, but normal platelet count and spleen size (erythrocythemic PV). Third, classic PV with increased RCM, erythrocytes above 6x1012/L, high hematocrit and one or more B criteria (classical PV, Table 1). Fourth, ET with a hypercellular trilinear myeloproliferation (ET.MGM Table 2) or unclassifiable or masked MPD with splenomegaly, normal hemoglobin and hematocrit, normal or slightly elevated platelet count [48-51].
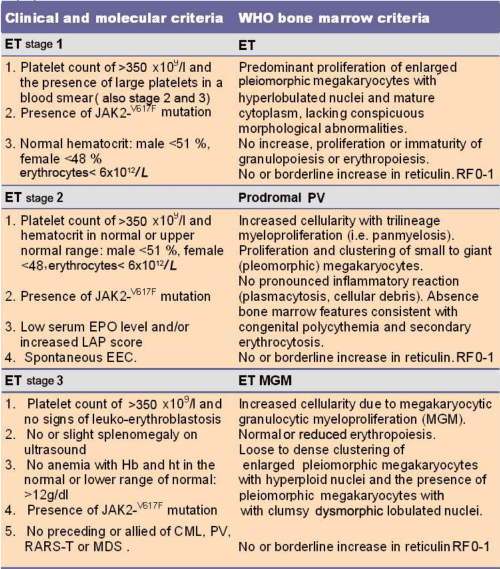
Table 2: 2008 ECMP criteria for the diagnosis of 3 phenotypes of JAK2V617F mutated Essential Thrombocythemia (ET): important to differentiate because the natural history may differ.
In the 2001 WHO criteria Thiele & Vardiman combined a characteristic PV histological bone marrow picture as a minor criterion with increased RCM as a major inclusion criterion for the diagnosis of PV [25], thereby excluding early thrombocythemic stage PV mimicking ET. Tefferi et al., revised the 1975 PVSG - 2001 WHO criteria into the 2007and 2008 WHO criteria into ET, PV and PMF [52]. 2008 WHO ET was defined by platelet count above 450x1012/L but did not distinguish the three stages of ET at the bone marrow level. The diagnosis of early thrombocythemic PV (hemoglobin <18.5 for men and <16.5 for women) and ET associated with prefibrotic PMF remained unclassifiable. The 2008 WHO criteria for PV arbitrarily exclude the early stages of PV and occult or masked PV just by the main inclusion criterion of a high hemoglobin level, and disregard increase of leukocytes, platelets and spleen size as typical features of trilinear PV. Simple tests like blood cell counts including platelets, leukocytes, hematocrit and erythrocytes and spleen size on echogram are not taken into account to distinguish the early thrombocythemic and erythrocythemic stages of PV from the overt trilinear polycythemic stage of classic PV as documented by bone marrow biopsy showing typical erythroid, granulocytic and megakaryocytic proliferation [52]. These shortcomings of the 2008 WHO diagnostic criteria for MPD will hamper prospective clinicopathological studies on natural history, staging and survival outcome of clinical and bone marrow defined ET, ET associated with prePMF (MF-0), and early prodromal PV, classical PV and masked PV. To overcome the shortcomings of the PVSG and WHO classifications of the MPDs, we here update the ECP and ECMP criteria for the diagnosis, classification and staging of ET, PV and PMGM [18,24,46,47].
Strenghts and limitations of PVSG-WHO criteria for the diagnosis of ET and PV
RCM measurement as a main PVSG-WHO inclusion criteria for the diagnosis of PV is cumbersome, time consuming, costly, and not specific for MPD. Increased RCM in patients with erythrocytosis does not distinguish early erythrocythemic PV from congenital polycythemia (CP) or secondary erythrocytosis (SE). In a consecutive cohort of 105 patients with WHO-defined PV, RCM had a sensitivity of 76% in the diagnosis of PV and a specificity of 79% in distinguishing PV and non-clonal polycythemia [53]. PV patients with increased RCM may have normal hemoglobin and hematocrit because of associated iron deficiency and/or significant splenomegaly, but erythrocyte count is always increased [54]. with values above 6x1012/L. WHO bone marrow criteria subclassify patients with increased RCM into patients with PV as a trilinear MPD and erythrocytosis (Table 4), either congenital, idiopathic or acquired [36,46-51]. RCM in patients with thrombocythemia, slight splenomegaly on echogram, and borderline serum EPO and hemoglobin levels does equally or even better distinguish between PVSG-defined ET from PV than bone marrow histopathology. In PV, RCM measurement data is found to be without additional diagnostic value, because all PV patients with increased red cell mass usually show a typical PV bone marrow histology and have erythrocyte counts above 6x1012/L [39,46,47].
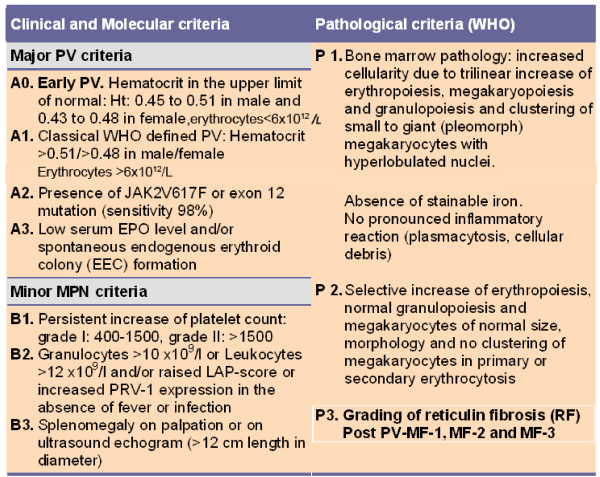
Table 3: The 2008 ECMP criteria for the diagnosis of Polycythemia Vera: PV.
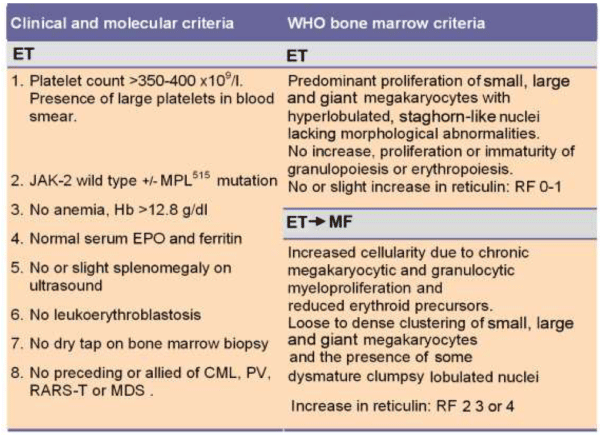
Table 4: The 2008 ECMP criteria for the diagnosis of MPL515 mutated normocellular ET.
Spontaneous EEC formation and low serum EPO levels are specific confirmative criteria for the diagnosis of PV, but have insufficient diagnostic sensitivity as isolated parameters to differentiate between PV, CP, CE, SE, ET and normal controls [55-60]. About 50% of PVSG defined ET patients show not only spontaneous EEC but also increased polycythemia rubra vera-1 (PRV-1) expression [61-65], together with low serum EPO levels [61,65,66], indicating that EEC/PRV-1-positive ET comprises a biologically distinct subgroup of ET patients reflecting early PV (“forme fruste” PV, Figure 2) that is at risk for progression to overt PV. In a study of 170 PVSG-defined ET patients, spontaneous EEC was seen in all 11 (6.5%), who later developed PV, and in 60% of 159 patients with stable ET during a median follow-up of 29 months (12-138 months) [67].
In a review of 120 cases with splanchnic vein thrombosis (Budd-Chiari syndrome 51, and portal/splenic and/or mesenteric vein thrombosis in 69), MPD was diagnosed in 80 with spontaneous EEC as a clue to MPD in 73 (61%). The diagnoses in the 80 MPD patients based on specific laboratory and bone marrow data were overt PV in 37 (31%), ET in 2, PMF in 2, and latent (masked) MPD in 39 (32.5%) [68]. At that time in 1997 we concluded that both spontaneous EEC and histopathology from bone marrow biopsy provide specific information as sensitive clues for the diagnosis of all variants of overt and latent myeloproliferative disorders [68]. Accounting clustered enlarged or giant megakaryocytes as diagnostic for MPD enabled French investigators to subsequently diagnose MPD in 46 out of 128 patients with splanchnic thrombosis either hepatic vein or portal vein thrombosis (Table 3) [69,70]. In this study, the sensitivity for the diagnosis of MPD was 63% for increased RCM, 52% for low serum EPO level, 72% for EEC formation, and 74% for splenomegaly, indicating the superiority of bone marrow histopathology to detect masked, early and overt stages of MPD as the underlying etiology in patients with idiopathic splanchnic vein thrombosis [69,70]. JAK2V617F mutation screening of 274 patients with splanchnic vein thrombosis, either hepatic, portal or mesenteric vein thrombosis in four recent studies appeared to be a specific clue to MPD in 86 cases (31%) [71-74]. Of these 86 JAK2V617 patients, 16 presented with overt MPD, and 70 (25.5%) had latent (masked) MPD of which 38 (overall 14%) developed MPD during follow-up when PVSG criteria are applied [71-74]. In two additional reports on patients with splanchnic vein thrombosis and masked MPD, the laboratory markers EEC, PRV-1 expression and low serum EPO were insensitive, whereas the combination of JAK2V617F mutation screening and bone marrow histology assessment was highly sensitive and specific to diagnose MPD [75,76].
The role of JAK2V617F mutation in the pathogenesis of trilinear MPD
The discovery of the JAK2V617F mutation by Vainchenkers and his team of MPD investigators [12,13], was immediately appreciated as an evolutionary event, and rapidly confirmed by several investigators (Figure 1) [26,27]. JAK2 plays an essential role in cytokine-induced signalling from receptors to the nucleus by several hematopoietic cytokines including erythropoietin (EPO), thrombopoietin (TPO), and granulocyte colony stimulating factor (G-CSF) [26,77-79]. The JAK2V617F mutation renders the receptors on mutated hematopoietic progenitor cells hypersensitive to these cytokines, thereby resulting in growth advantage of the mutated aver the non-mutated trilinear hematopopoietic cells present in the bone marrow. The JAK2V617F mutation is detectable in CD34+ hematopoietic bone marrow cells, erythroblasts, in cells of spontaneous EEC, blood platelets and granulocytes (Figure 1) [77-79]. Applying allele-specific polymerase chain reaction (PCR) analysis in PVSG-defined MPD patients, a high frequency of the JAK2V617F mutation of 95% (92-97%) is described in PV, and a lower frequency of 53% (49-57%) in ET and 52% (44-55%) in CIMF [26,80]. Only 3 to 4% of ET, 24 to 27% of PV and 6 to 18% of CIMF patients are homozygous for the JAK2V617F mutation [26,80].
Based on animal studies and different mutation states of JAK2V617F in MPD patients, two hypotheses have been proposed to explain why three different phenotypes of MPD are caused by the same JAK2V617Fmutation: the “dosage” hypothesis and the “additional events” hypothesis [78-80]. According to the “dosage” hypothesis the level and duration of JAK2V617F directly contribute to the phenotypic diversity of trilinear MPDs [78,79]. This hypothesis is based on different densities of TPO receptors (TPOR or MPL)) and EPO receptors (EPOR) on hematopoietic progenitor cells and on differences of response of TPOR and EPOR to various levels of JAK2V617F activity [78,79]. TPOR/MPL is expressed at high levels in megakaryocytic cells where it controls physiological TPO levels. It is possible that activation of a few TPO receptors by low levels of JAK2V617F (heterozygous) is sufficient to send a signal to megakaryocytic cells. A slight increase in numbers of mutated large (giant) megakaryocytes and platelets (about 50 to 100 x109/l mutated platelets) might be sufficient to produce platelet-mediated microvascular circulation disturbances (Figure 1) [26,80]. Conversely, EPOR is expressed at low levels on hematopoietic progenitor cells and therefore high levels of JAK2V617F may be required to activate EPOR and generate a PV-like phenotype [26,80]. Sustained high levels of JAK2V617F during long-term follow-up subsequently may lead to a high level activation of activation of EPOR and GCSF receptor (GCSFR) leading to extramedullary hematopoiesis (splenomegaly) and cytokine mediated secondary myelofibrosis [78,79]. The percentage of JAK2V617F positivity and progression from heterozygous to homozygous is strongly correlated with increased PRV-1 expression in granulocytes, with the ability to form spontaneous EEC formation and with progressive post-PV myelofibrosis [64,81]. Scott et al., showed that BFU-e colonies are already homozygous for the JAK2V617F mutation in PV patients with a heterozygous pattern of JAK2V617F in their peripheral blood granulocytes [82]. In contrast, the BFU-E colonies from heterozygous patients with ET did not contain a subpopulation of JAK2V617F homozygous cells (Figures 1,2) [82]. French investigators studied a large group of JAK2V617F positive PV (N=159, 36% homozygous) and ET (N=147, 4% homozygous), and genotyped BFU-E colonies in in 20 PV and 6 ET patients [83]. They showed that JAK2V617F positive ET patients usually harbour heterozygous BFU-E clones, some PV patients have a purely heterozygous profile, and most PV patients have a mixture of heterozygous and homozygous BFU-E clones [83]. Mutated erythroid progenitors are more sensitive to EPO than normal progenitors, and most homozygous progenitors are EPO independent. In this cohort of 306 JAK2V617F positive MPD patients, PV was associated with significantly lower platelet counts and higher hematocrit and granulocyte values than ET patient. The highest platelet count was associated with low JAK2V617F levels in PV, whereas high JAKV617F levels correlated with high hemoglobin and high granulocyte counts in ET [83]. A recent study detected JAK2V617F in 75% of ET (n=60) and in 97% of PV patients (n=62), whereas allelic ratios exceeding 50% JAK2V617F indicating homozygosity were found in 70% of PV at diagnosis but never in ET [84]. Transition from heterozygosity to homozygosity for the JAK2V617F mutation represents a very important step in the progression from early to classic PV and subsequent post-PV myelofibrosis [81]. Comparing JAK2V617F heterozygous and homozygous PV patients showed that homozygote JAK2V617F PV patients displayed significantly higher hemoglobin at time of diagnosis, increased incidence of pruritus, higher PRV-1 expression in granulocytes, and a higher rate of fibrotic transformation [84]. Sex appears to be a powerful genetic background modifier in JAK2V617F-positive MPDs as ET is more common in females and PV in males.
Mechanisms other than mitotic recombination such as duplication of the mutated allele is observed in a proportion of PV and MF patients displaying a gain of 9p, mostly due to trisomy 9 [85-87]. Campbell et al., reported that the JAK2V617F mutation was associated with trisomy 9 with all 10 MPD patients investigated and was found in 28 of 29 MPD patients (PV, ET or MF) with a 20q deletion [87]. Scott et al., identified JAK2 exon 12 mutations in 10 erythrocytosis patients with increased red cell mass but no JAK2V617F, of which according to PVSG criteria 6 could be diagnosed as PV and 4 as idiopathic erythrocytosis [88]. Pre-treatment bone marrow biopsies in 5 patients carrying a JAK2 exon 12 mutation showed a characteristic pattern of erythroid hyperplasia without morphological abnormalities of the megakaryocyte or granulocyte lineages [88]. Therefore, an overlap between “dosage” and “additional molecular events” hypotheses is very likely in patients with trilinear PV [78,79].
Acquired MPL gain of function mutations as the cause of normocellular ET
The JAK2 kinase activity in MPDs is not only dependent on the amount of heterozygous and homozygous JAK2V617F mutant protein, but may also be influenced by the various steps upstream or downstream the signalling pathways including MPL, JAK2, STAT-3. This has been demonstrated in animal models overexpressing c-MPL [89]. MPL transgenic mice manifested with typical features of ET with a fourfold increase of platelet count, increased colony formation of megakaryocytes, and increase of clustered enlarged megakaryocytes in the bone marrow. The ET animals appeared healthy, had a very slight decrease of hematocrit (0.39 versus 0.42 in controls) despite an increase of bone marrow EEC, and survived normally with no evidence of myelofibrosis in the bone marrow [89]. The acquired MPLW515L and MPLW515K gain of function mutations have recently been discovered as the underlying etiology in ET patients [90,91]. MPL515 is a somatic activating mutation in the transmembrane domain of MPL, the thrombopoietin receptor (TPOR), which was found in 4 of 45 (9%) of JAK2 wild type myelofibrosis patients [81], and in about 10% of MF patients in France [77]. In a mouse model transplant assay, overexpression of the MPLW515L mutation (100%) resulted in a fully penetrant MPD characterized by marked thrombocytosis and leukocytosis with no evidence of PV, but with increase and clustering of enlarged dysmorphic megakaryocytes in the bone marrow, myelofibrosis and marked splenomegaly due to extra medullary hemopoiesis consistent with MF [90]. Screening of 1182 PVSG-defined MPD patients (318 ET, 242 PV, and 290 IMF) and 64 controls for MPL515 mutations resulted in the detection of MPL mutations either MPLW515L (n=17) or MPLW515K (n=5) in 20 MPD patients (de novo CIMF in 4%, ET in 4 = 1%, and post-ET myelofibrosis in 1), but not in the 242 PV patients and controls [91]. Six cases carried both MPLW515L and JAK2V617F alleles indicating that these alleles have functional complementation in CIMF. MPL515 mutation related myelofibrosis may represent a distinct entity of JAK2 wild type MF without features of PV and distinct from JAK2V617F trilinear MPD. These observations are in line with the “additional molecular events” hypothesis, indicating that alternative or additional molecular abnormalities, like JAK2V617 mutation alone, combinations of JAKV617F and MPL515 mutations or other combinations of still unknown mutations contribute to or modify the MPD phenotypes.
Hannover bone marrow classification for the MPDs ET, PV and PMGM
In 1990 Georgii et al proposed the Hannover Classification to clearly distinguished three distinct primary MPDs ET, PV and CMGM/PMGM (Figure 2) [18]. With the improvement of bone marrow biopsy and tissue processing in the 1980s, Georgii defined the pathological features of ET, PV and chronic megakaryocytic granulocytic myeloproliferation (CMGM) on bone marrow histopathological morphology [18,100,101,106]. ET was defined by persistent increase of platelets in excess of 400 x109/l without the Ph+ chromosome together with monolinear proliferation of mature enlarged megakaryocytes in the bone marrow with normal cellularity, normal erythropoiesis and normal granulopoiesis (Figure 2) [18,24-26,33,100-104]. PV was defined as a trilineage proliferation of megakaryopoiesis, erythropoiesis and granulopoiesis in which the erythropoiesis was most prominent together with variable degrees of increased platelets, erythrocytes and granulocytes in the peripheral blood in the absence of the Ph+ chromosome [14-18,100-104]. Georgii regarded myelofibrosis (MF) as a reactive feature secondary to progressive disease [18] seen in CMGM, PV and CML [18,100-104,106]. Georgii reasoned that the terms agnogenic myloid metaplasia (AMM) or chronic idiopathic myelofibrosis (CIMF) used by Thiele and Vardiman in the 2001 WHO classification lack accuracy since they are applied to both the prefibrotic hypercellular and advanced fibrotic stages [4-17,102-105]. The diagnosis of prefibrotic PMGM [18,100,101] is based on identical bone marrow criteria: 1) the presence of large megakaryocytes with immature cytoplasm and immature cloud-like nuclei not seen in ET and PV, 2) increased granulopoiesis but never disturbed in maturation and 3) usually relatively decreased erythropoiesis (Figure 2). As prefibrotic CMGM is typically featured by thrombocythemia associated with typical primary megakaryocytic and granulocytic myeloproliferation (PMGM) in the complete absence of myelofibrosis (MF) Michiels preferred to the term primary megakaryocytic granulocytic myeloproliferation (PMGM) simple because myelofibrosis (MF) is not a disease but a secondary complication of MPD (Table 5) [106,111] Consequently, Michiels & De Raeve followed the Hannover BM classification to change the PVSG-WHO classifications into the ECP (Table 1), [46,47] and the 2008 ECMP [112] criteria for the diagnosis of the Ph-negative MPDs to describe the full spectrum of WHO bone marrow features for ET, early prodromal PV, classical PV, Masked PV and ET associated with PMGM (Tables 2-5).
| Table 5. Grading of myelofibrosis (MF) according to Baumeister, USA137, Manoharan, UK138, and the European consensus (EC) agreement in 2005 by Thiele et al139 in bone marrow biopsies of patients with a chronic myeloproliferative disorder. | |||
| USA137 Subjective |
UK138 Subjective |
EC grading139 Descriptive |
EC139 |
| MF 1 | 1+ |
Scattered linear fine fibers with no intersections (cross-overs) and rare course reticukin fibers |
MF 0 Prefibrotic |
| MF 2 | 2+ 3+ |
Loose network of reticulin with intersections, especially in perivascular areas, no collagenization |
MF 1 Early fibrotic |
| MF 3 | 4+ | Diffuse and dense increase in reticulin with extensive intersections, occasionally only focal bundles of collagen and/or focal osteosclerosis | MF 2 Fibrotic |
| MF 4 | Diffuse and dense increased in reticulin with extensive interactions with coarse bundles of collagen, often associated with significant osteosclerosis | MF 3 Sclerotic |
|
Grading of myelofibrosis in myeloproliferative disorders
Myelofibrosis (MF) itself is not a disease because reticulin and collagen fibrosis are produced by polyclonal fibroblasts in response to cytokines released from the clonal granulocytic and megakaryocytic proliferative cells in both PV and MF (Table 5) [18,136]. Transformation to myelofibrosis is rare in ET and does occur in about one third of PV [100,101,106,107,115]. The grading of the Baumeister scoring system of MF was developed on aspirated bone marrow samples, is poorly defined and not reproducible for the proper grading of myelofibrosis in bone marrow biopsies by pathologist (Table 3) [137]. The Manoharan system used silver stain according to Gordon and Sweet scored the degree of reticulin in bone marrow biopsy in a completely different way (Table 5) [138]. A scoring system based on morphometric analysis (point intersection with an ocular grid) and quality of fibers (reticulin and collagen fibers) and the bone marrow fiber density (fine or course reticulin and some or course bundles of collagen) has been proposed by German institutes of pathology [18,100-108]. All these different scoring systems for MF use different criteria for grading of reticulin and collagen, are subjective and not comparable by lack of strict criteria. A panel of experienced European pathologists and an USA expert reached a consensus on how to grade bone fibrosis in bone marrow biopsies of patients with CIMF or PV [139]. Grading of MF was simplified by using four easily reproducible categories that included differentiation between reticulin and collagen [139]. According to defined standardized semiquantitative grading of reticulin and collagen fibrosis in the bone marrow, MF can reliably be graded at the pathological bone marrow level as 0 in prefibrotic, as 1 in early fibrotic, as 2 in classical fibrotic and as 3 in classical sclerotic MF (Table 5) [139].
Definition of ECMP criteria for the diagnosis and classification of MPD
The ECMP criteria combine the major features from the PVSG-WHO classifications with Hannover BM features and incorporates specific laboratory markers (including EEC, serum EPO, JAK2V617F, MPL515) for the diagnosis and staging of the three prefibrotic myeloproliferative neoplasms (MPNs) ET, PV and PMGM (Tables 1, 2, 3 and 4, Figures 3 and 4) [112]. For the diagnosis of the MPN trephine bone marrow biopsy specimens should be embedded in paraffin or plastic, which have both their technical limitations. Paraffin requires decalcification with EDTA (preferable, allows reasonable DNA quality) or acid electrolysis. The specimens should have at least 4 evaluable bone marrow spaces with hematopoiesis. Recommended stains include: hematoxylin and eosin (H&E), Giemsa (3 µm sections), periodic acid-Schiff (PAS); Perls for estimation of hemosiderin content; chloro-acetate esterase (Leder) for identification of granulocytic differentiation; silver stain for reticulin; and trichrome-Masson for collagen staining. Immunostains of paraffin embedded specimens should include glycophorine C for erythropoiesis, myeloperoxydase for granulopoiesis, CD42b, CD61 or FVIII- related antigen for megakaryocytes; CD34 for CD34-positive hematopoietic progenitor cells or blasts. Regarding MPD, clinicians want to receive from their pathologist a classifying diagnosis either ET, PV or ET associated with PMGM in newly diagnosed MPD patients based on a detailed report according to the Cologne bone marrow evaluation form [46,47,111,112].
ET. According to 2008 ECMP criteria, increase and loose clustering of enlarged mature megakaryocytes with hyper-lobulated nuclei in a normocellular bone marrow and platelet count >400 x109/l represent the hallmark of ET. In ET there is no proliferation or immaturity of granulopoiesis or erythropoiesis. In congenital and acquired erythrocytosis and in reactive thrombocytosis the megakaryocytes are of normal size and morphology and there is no tendency to cluster (Figure 3). A typical histopathological ET picture of the bone marrow excludes RT and distinguishes ET from early PV and clearly defines ET associated with PMGM as clearly distinct from thrombocythemias associated with atypical MPD, MDS, refractory anemia with increased ringed sideroblasts (RARS-T) or Ph+–positive thrombocythemia in CML.
PV. The characteristic increase and clustering of small and enlarged pleomorphic megakaryocytes and increased erythropoiesis with increased granulopoiesis and increased cellularity (according to age) are the diagnostic characteristics of classic PV with increased erythrocytes above 6x1012/L and hematocrit above 0.51 combined with low serum EPO and JAK2V617F mutation distinguishing it from congenital and secondary erythrocytosis. A typical histological PV picture with moderately increased bone marrow cellularity is seen in patients with early PV mimicking ET or “forme fruste” PV featured by platelet count >400x109/l and hematocrit <0.51, low serum EPO and/or the presence of the JAK2V617F mutation. A typical histological PV bone marrow picture is also seen in the early erythrocythemic stage 1 PV (hematocrit >0.51, platelet count <400 x109/l, normal spleen, low serum EPO) with the presence of the JAK2V617F mutation.
ET associated with PMGM (MF-0): According to Hannover [18], Cologne [27], WHO [25] and ECMP [112] classifications, ET associated with PMGM is characterized by hypercellularity of the bone marrow due to increased granulopoiesis, relative decrease of erythropoiesis and the presence of dense clusters of immature megakaryocytes with bulky nuclei showing lobuli and irregular roundish forms (so-called cloud-like nuclei), which are almost never seen in ET and PV (Table 6). The degree of dysmegakaryopoiesis in ET associated with PMGM (MF-0) may range from atypical slight maturation defect of megakaryocytes with no cloud-like nuclei to typical maturation defects of megakaryocytes with typical cloud-like nuclei or a mixture of both (Table 6). The risk of ET associated with PMGM (MF-0) to progress to early MF-1 and subsequent CMF-2/3 with extramedullary hematopoiesis is related to the degree of hypercellularity and on the degree of maturation defects of megakaryopoiesis [18,107,108].
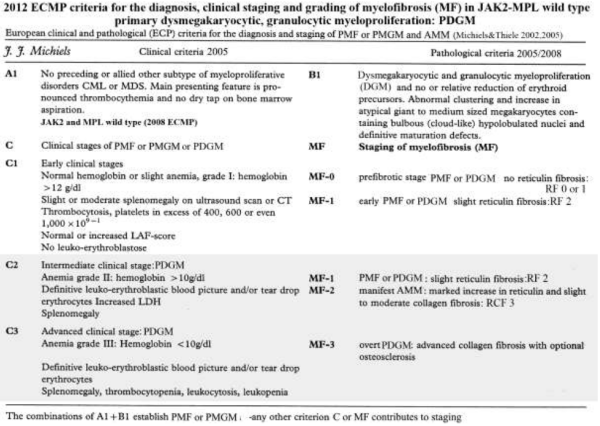
Table 6:
High quality histological bone marrow preparations in the hands of experienced pathologists are required to achieve discrimination of ET associated with PMGM (MF-0) from true ET and PV in about 85 to 90% of the cases [14-18,100-108]. There are no studies that have examined the concordance between a number of pathologists who have used characteristic histological bone marrow features to assign cases to the prefibrotic stages of true ET, PV and ET associated with PMGM (MF-0) without knowledge of the biological MPD markers and the clinical findings except age. Data on the very long-term natural history of patients with ECMP defined ET, prodromal PV and ET associated with PMGM as derived from large scale prospective studies are lacking. We do not really know whether the early prodromal stages of PV patients are at no, low or high risk of progression to post-PV myelofibrosis or whether heterozygous JAK2V617F mutated ET never progresses to fibrotic MF as it is claimed. Masked PV featured by trilinear hypercellular bone marrow with increased erythropoiesis, megakaryopiesis and granulopoiesis (EMG) is associated with variable degrees of thrombocythemia, leukocytosis, increased LAP-score and moderate to pronounced splenomegaly (labelled as inapparent PV by Laky et al.).
The 2008 ECMP criteria separate ET patients into three phenotypes of prefibotic MPNs at the bone marrow level (Table 1). ET, prodromal PV mimicking ET and ET associated with atypical and typical PMGM (MF-0) without features of PV and absence of leuko-erytroblastosis in the peripheral blood [107-112]. These three ECMP defined ET phenotypes do not differ significantly with regard to peripheral blood features, thrombocythemia related clinical presentation or laboratory findings [107,108]. Therefore, patients with normocellular ET and early PV and ET associated with PMGM (MF-0) are to be treated equally based on clinical risk stratification for thrombotic and bleeding complications [80]. Progression of normocellular ET into myelofibrosis was not seen five years after diagnosis in two studies, but data on very long-term follow-up are lacking [108,114,115-117].
Diagnostic work-up of patients with ET in various MPNs
Clinical manifestations of thrombocythemia in masked and overt MPNs consist of microvascular circulation disturbances including atypical and typical TIAs, ocular ischemic attacks, erythromelalgia, and splanchnic vein thrombosis [92-95]. Sustained increase of platelet counts (>400 x109/l) associated with slight splenomegaly on echogram (>12cm), increased leukocytes (>12 x109/l), or LAP score with normal ESR is highly suspicious of WHO-ECMP defined ET or thrombocythemia in various MPN with the absence of any cause for reactive thrombocytosis (Figure 2). The presence of giant platelets in a peripheral blood smear is indicative for MPN. The presence of numerous abnormal enlarged or giant mature or dysmorphic megakaryocytes and clustered larged megakaryocytes in bone marrow biopsy are the pathognomonic clues to the diagnosis of prefibrotic MPN. The diagnostic work-up of patients with thrombocythemia in various MPDs ET, PV, ET associated with PMGM (MF-0 or MF-1) is based on bone marrow histology findings. These include:
Thrombocythemia patients should fulfil the peripheral blood (clinical) criteria for the diagnosis of thrombocythemia irrespective of bone marrow features (Figure 3).
JAK2V617F mutation screening as a first intention diagnostic test is very helpful in the diagnostic work-up of patients with suspected thrombocythemia in various MPDs, but only half of ET and MF patients carry this mutation (sensitivity 50-60%).
Pretreatment bone marrow biopsy will allow clinicians and pathologists to diagnose the early stages of thrombocythemia in various MPNs irrespective of JAK2V617F mutation status. The 2008 ECMP criteria classify JAK2V617F mutated MPN into 3 phenotypes or stages (Table 2, Figure 3), normocellular ET phenotype 1; ET phenotype 2 with features of PV (prodromal PV); hypercellular ET associated with megakaryocytic granulocytic myeloproliferation (prefibrotic ET-MGM or masked PV) without features of leukoerythrocytosis and extramedullary hematopoiesis.
The 2008 ECMP criteria distinguish thrombocythemia in various MPDs from thrombocythemia associated with Ph1-chromosome and BRC/ABL positive chronic myeloid leukemia (CML) and ET [118] or myelodysplastic syndromes (MDS) including the so-called 5q-syndrome, which clearly differs from refractory anemia with ringed sideroblasts and significant thrombocytosis (RARS-T) (Figure 3) [119-121]. Among 9 RARS-T patients, 6 showed the presence of JAK2V617F mutation [121,122].
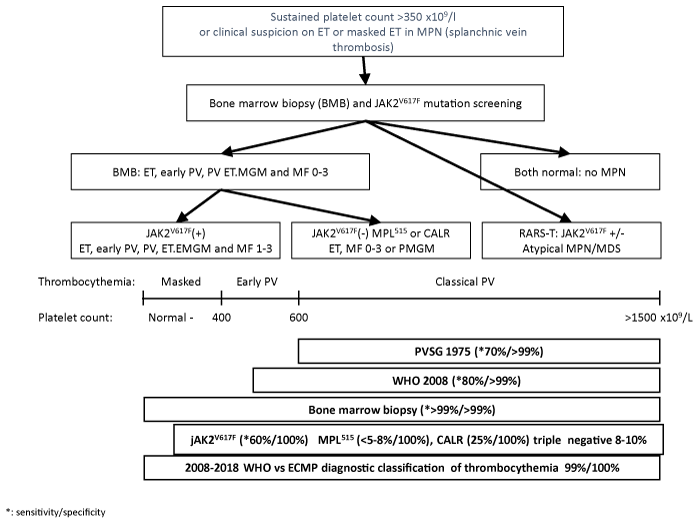
Figure 3: Algorithm for diagnostic work-up for patients with suspected thrombocythemia as the presenting feature of ET, early PV, prefibrotic CIMF-0 (ET-MGM), early fibrotic CIMF-1 or refractory anemia with increased ringed sideroblasts (RARS-T) [112]. For explanation see text.
As compared to JAK2 wild type ET, JAK2V617F positive ET is characterized by higher values for hemoglobin, hematocrit, neutrophil counts, LAP score, by lower values for serum EPO levels, serum ferritin and MCV, and by increased cellularity of the bone marrow in biopsy material [113,114], indicating early thrombocythemic PV mimicking ET (“forme fruste” PV, stage 1 PV, Table 1, Figure 3) [26,111,112]. JAK2 wild type ET patients represent a distinct category who had significantly higher platelet counts, normal serum EPO levels, a typical bone marrow picture of ET, no features of early PV, and are at lower risk for the development of thrombotic complications [122,123]. These data are in line with the fact that JAK2V617 positive and JAK2 wild type ET patients at diagnosis represent two distinct entities with a related pathophysiology in the JAK-2/STAT signalling pathway but different molecular etiology.
Diagnostic work-up of patients with polycythemia vera: PV versus erythrocytoses
PV patients frequently present with headache, TIAs, erythromelalgia [92-95], splanchnic vein thrombosis [68-76], and microcytosis of erythrocytes due to iron deficiency [123,124]. Characteristic PV features include increased hematocrit (>0.51), increased erythrocytes (>6 x1012/l), slight splenomegaly, increased leukocytes (>12 x109/l) or LAP score with normal ESR, increased platelets (>400 x109/l) (Table 2, Figure 4). PV patients usually show the presence of large platelet in peripheral blood smear. Patients with congenital erythrocytosis with a gain of function mutation in the EPOR or acquired erythrocytosis lack the clinical, laboratory, molecular and bone marrow features of MPD and are usually asymptomatic [125,126]. The detection of JAK2V617F in granulocytes with sensitive PCR techniques plays a key-role as a first intention diagnostic test for erythrocytosis, because it simplifies the diagnostic work-up of PV (Figure 4) [112,127,128]. In the context of erythrocytosis (hematocrit >0.51 in males and >0.48 in females) the presence of the JAK2V617F mutation has a sensitivity of 95% and positive predictive value of 100% for the diagnosis of PV, and excludes congenital and secondary erythrocytosis (Figure 4) [112,127,128]. EEC and low serum EPO significantly contribute but are not sensitive enough to diagnose the broad spectrum of PV phenotypes [55,56,129-133].
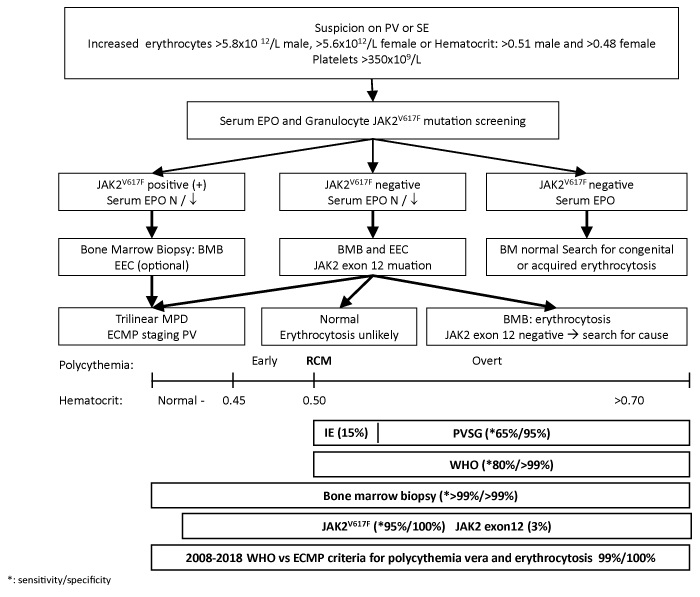
Figure 4: Algorithm for diagnostic work-up of patients with suspected polycythemia vera versus primary or secondary erythrocytosis [112]. For explanation see text.
In contrast to increased serum EPO levels in cases secondary erythrocytosis, generally below-normal serum EPO levels have been found in early and overt PV [60,129-133]. The decrease in serum EPO levels is appropriate for the level of hematocrit due to autonomous increased erythropoietin in the bone marrow. EEC is time consuming and difficult to establish in many (non-specialized) laboratories, and not sensitive enough to distinguish PV from primary or secondary erythrocytosis [59]. In contrast, bone marrow histology clearly differentiates trilinear hypercellularity in PV from an isolated increase of erythropoiesis in congenital polycythemia and secondary erythrocytosis (Figure 3). Clinicians and pathologists tend to replace EEC by the wide spread available bone marrow histology assessment as a gold standard for the diagnosis of masked, overt and advanced JAK2V617F mutated and exon 12 mutated PV [68-70,88,134]. Differential diagnosis of JAK2 wild type classic PV, early erythrocythemic PV and idiopathic erythrocytosis may be problematic [88,124]. Red cell mass measurement does not distinguish myeloproliferative PV from primary and secondary erythrocytoses. The combined use of molecular screening and bone marrow histology has a sensitivity and sensitivity of 100% to distinguish JAK2 wild type primary and secondary erythrocytoses from JAK2V617F mutated, JAK2 exon 12 mutated IE and PV.
The diagnostic role of JAK2 mutation detection and bone marrow histology in MPN
Bone marrow biopsy significantly contributes to the diagnostic differentiation between true heterozygous ET and early homozygous PV. Bock et al., evaluated the JAK2V617F mutation using PCR techniques in bone marrow cells derived from bone marrow trephine biopsies from 79 MPD patients classified according to PVSG-WHO classification ET, PV or PMF [141]. The JAK2V617F mutation was found in 90% of PV (n=29), 22% in PMF (MF-0, n=18), 60% in advanced PMF (n=20), and 27% in normocellular ET (n=15), but not in CML (n=5), acute leukemia (n=20) secondary erythrocytosis (n=10, or normal bone marrow (n=10) [141]. The JAK2V617F mutation occurred at a lower frequency in normocellular ET never exceeding 50% of alleles indicating heterozygosity, and exceeding 50% of alleles indicating homozygosity in PV and fibrotic PMF. Bock et al., evaluated the JAK2V617F status from trephine bone marrow biopsies in a second series of 64 patients with PMF. The frequency of JAK2V617F mutation was 45% (9% homozygous, 36% heterozygous) in early hypercellular prePMF MF-0 or MF1 (n-31) and 53% (13% homozygous and 40% heterozygous) in advanced PMF MF-2/3 (n-33) [142]. Horn et al., studied 152 paraffin-embedded trephine bone marrow biopsies from patients with MPD diagnosed according to PVSG-WHO criteria for the presence of the JAK2V617F mutation using PCR techniques [143]. Only 6 of 152 samples were not evaluable because of poor DNA quality. The JAK2V617F mutation was detected in 27 of 28 (96%) cases of PV, 17 of 23 (74%) cases of normocellular ET, 28 of 45 (75%) of PMF with MF-0 to 3, in 8 of 12 (75%) cases of MPD unclassifiable or with MDS/MPD syndrome, but not in Ph-chromosome positive CML (n=4), secondary erythrocytosis or reactive thrombocytosis (n=15) and controls (n=19). Data on JAK2V617F mutation burden and the diagnostic differentiation between heterozygous or homozygous for the JAK2 mutation are lacking in this study [143].
The JAK2V617F mutation is very sensitive to detect the early latent (masked) stages of ET and PV particularly in those young MPD patients who present with migraine-like headache, atypical TIAs, Budd-Chiari syndrome or splanchnic vein thrombosis. The combined use of the recently discovered JAK2 and MPL mutations with bone marrow histology appears to be pathognomonic and powerful diagnostic tools to identify the latent (masked), early and overt MPDs [112]. Within the JAK2V617F-positive MPDs, three main clinicopathological subtypes can easily be distinguished. ET is an indolent slow onset myeloproliferation of mature enlarged megakaryocytes. The combination of increased platelet count, EEC and/or low serum EPO at hematocrit below 0.50 is consistent with early thrombocythemic prodromal PV. The combination of JAK2V617 mutation, EEC, low serum EPO and increased erythrocytes above 6x1012/L and hematocrit above 051 in males, above 0.48 in females, but normal platelet and leukocyte counts and normal spleen size is consistent with early erythrocythemic PV, formerly categorized as idiopathic erythrocytosis. Early thrombocythemic PV, early erythrocythemic PV, and classic PV are caused by JAK2V617F mutation in 95%. JAK2 wild type PV is rare (5%) and frequently carries a heterozygous JAK2 exon 12 mutation [88]. PV patients “heterozygous for the JAK2 mutation in granulocytes” are usually homozygous for JAK2V617F in BFU-E at the bone marrow level [77], which may readily explain the rapid onset and progressive nature of the disease. JAK2V617F positive MF may be regarded as a variant of PV with predominance of granulocytic myeloproliferation and no or relative decrease of erythropoiesis. The degree of dysmegakaryopoiesis in overt and advanced PV, ET associated with PMGM (MF-0 and MF-1) seems to be associated with increased risk of progression to myelofibrosis [112,144]. Evidences accumulate that JAK2 wild type normocellular ET and PMF lack specific PV laboratory and pathological features at diagnosis and during follow-up. This has been demonstrated for MPL515 mutated MPD (ET/MF) both in humans and in animal models [90,91]. Whether the finding of very low levels of the JAK2V617F mutation around 1% in controls and in cases with suspected MPD is of relevance remains elusive [145-147]. This issue can only be solved by the combined use of WHO-ECMP markers to clearly define and document MPN patients [112,144]. Recently we could define JAK2/MPL wild type hypercellular ET associated with primary megakaryocytic granulycytic myeloproliferation (PMGM) as a third distinct MPN entity caused by the CALR mutation al (Figure 5) [148-152]. The flexible use of WHO-ECMP criteria should serve as pathognomonic diagnostic clues to each of the prefibrotic MPDs, will distinguish early and overt PV from primary or secondary erythrocytosis, and can be applied to document the natural history of myeloproliferative and fibrotic disease in ET, PV and MF patients. Further improvement of the WHO-ECMP classification of prefibrotic and fibrotic MPDs is predicted to have an impact on natural history with regard to transition into overt PV or progressive MF.
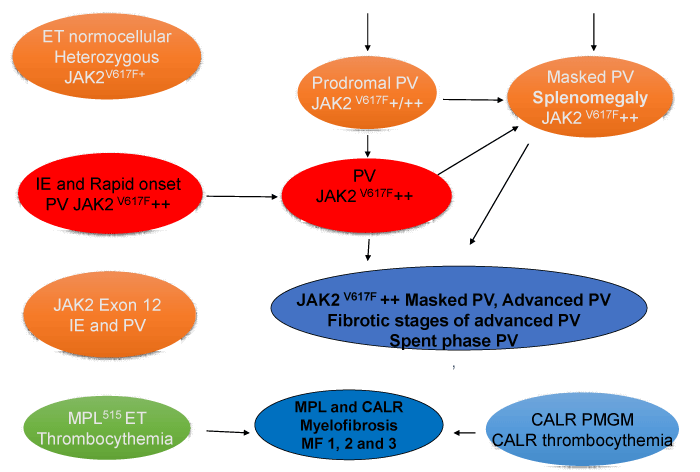
Figure 5: 2018 Clinical Laboratory Molecular and Pathological (2018 CLMP) translational states of four distinct JAK2/MPL/CALR mutated Myeloproliferative Neoplasms: CLMP classification of the MPNs according to Michiels & De Raeve.
References
- Heuck G. Two cases of leukemia with peculiar blood resp. Bone marrow findings. Virch Archiv. 1879; 78: 475-496. http://tinyurl.com/y3nj4d9a
- Vaquez MH. On a special form of cyanosis accompanied by excessive and persistent hyperglobulism. Minutes of meetings of the Society of Biology. 1892; 44: 384-388.
- Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951; 6: 372-375. Ref.: http://tinyurl.com/y3jaylqy
- Nowell PC, Hungerford DA. A minute chromosome in human chronic, granulocytic leukemia. Science. 1960; 142: 1497. http://tinyurl.com/y643rqmp
- Rowley J. A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence Giemsa staining. Nature. 1973; 243: 290-291. Ref.: http://tinyurl.com/y6q2cge5
- De Klein A, Van Kessel AG, Grosveld GG, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukemia. Nature. 1982; 300: 765-767. Ref.: http://tinyurl.com/yyt56e7j
- Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of BCR/ABL oncogene products. Science. 1990; 247: 1079-1082. Ref.: http://tinyurl.com/y27g6qbd
- Kelliber MA, McLaughin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous-like syndrome in mice with v-abl and BCR/ABL. Proc Nat Sci. 1990; 87: 6649-6653. Ref.: http://tinyurl.com/yya3hopy
- Shephard PCA, Ganesan TS, Galton DAG. Haematological classification of the chronic myeloid leukemias. Baillière’s Clin Haematol. 1987; 1: 887-906. Ref.: http://tinyurl.com/y27glcpu
- Michiels JJ, Prins ME, Hagemeijer A, Brederoo P, Van der Meulen J, et al. Philadelphia chromosome-positive thrombocythemia and megakaryoblast leukemia. Am J Clin Pathol. 1987; 88: 645-652. Ref.: http://tinyurl.com/y4wyaeaa
- Dameshek W. Physiopathology and course of polycythemia vera as related to therapy. J Am Med Ass. 1950; 142: 790-797. Ref.: http://tinyurl.com/y2b5pqa2
- James C, Ugo V, Le Couedic PF, Staerk J, Delhommeau F, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythemia vera. Nature. 2005; 434: 1144-1148. Ref.: http://tinyurl.com/yxnm998a
- James C, Ugo V, Casadevall N, Constantinescu SN, Vainchenker W. A JAK2 mutation in myeloproliferative disorders: pathogenesis and therapeutic and scientific prospects. Trends Mol Med. 2005; 11: 546-554. Ref.: http://tinyurl.com/y66ty8n8
- Thiele J, Zankovich R, Schneider G, Kremer B, Fischer R, et al. Primary (essential) thrombocythemia versus polycythemia rubra vera. A histomorphometric analysis of bone marrow features in trephine biopsies. Analyt Quat Cytol Histol. 1988; 10: 375-382. Ref.: http://tinyurl.com/y4oesnkw
- Thiele J, Schneider G, Hoeppner B, Wienhold S, Zankovich R, et al. Histomorphometry of bone marrow biopsies in chronic myeloproliferative disorders with associated thrombocythosis – features of significance for the diagnosis of primary (essential) thrombocythemia. Virch Arch A Path Anat. 1988; 413: 407-417. Ref.: http://tinyurl.com/y3lqtokm
- Thiele J, Zankovich R, Steinberg T, Kremer B, Fischer R, et al. Primary (essential) thrombocythemia versus hyperplastic stages of agnogenic myeloid metaplasia with thrombocytosis: a critical evaluation of clinical and histomorphological data. Acta Haematol. 1989; 81: 192-202. Ref.: http://tinyurl.com/y5vevoy7
- Thiele J, Zankovich R, Steinberg T, Fischer R, Diehl V. Agnogenic myeloid metaplasia (AMM) – correlation of bone marrow lesions with laboratory data: a longitudinal clinicopathological study on 114 patients. Hematol Oncol. 1989; 7: 327-343. Ref.: http://tinyurl.com/yyusm65l
- Georgii A, Vykoupil KF, Buhr Th, Choritz H, Doehler U, et al. Chronic myeloproliferative disorders in bone marrow biopsies. Path Res Pract. 1990; 186: 3-27. Ref.: http://tinyurl.com/y4v9x6t2
- Wasserman LR. The management of polycythemia vera. Br J Haematol. 1971; 21: 371-376. Ref.: http://tinyurl.com/y2vsn9h3
- Berlin NI. Diagnosis and classification of the polycythemias. Sem Hematol. 1975; 12: 339-351. Ref.: http://tinyurl.com/yyqkheob
- Laszlo J. Myeloproliferative disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD and hemorrhagic thrombocythemia. Semin Hematol. 1975; 12: 409-432. Ref.: http://tinyurl.com/y68mreem
- Murphy S, Iland H, Rosenthal D, Laszlo J. Essential thrombocythemia: An interim report from the Polycythemia Vera Study Group. Semin Hematol. 1986; 23: 177-182. Ref.: http://tinyurl.com/y4pwch4j
- Murphy S, Peterson P, Iland H, Laszlo J. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Sem Hematol. 1997; 34: 29-39. Ref.: http://tinyurl.com/y6fmr7pb
- Michiels JJ. Diagnostic criteria of the myeloproliferative4 disorders (MPD): essential thrombocythemia, polycythemia vera, and chronic megakaryocytic granulocytic metaplasia. Neth J Med. 1997; 51: 57-64. Ref.: http://tinyurl.com/y6s2az6b
- Jaffe S, Harris NL, Stein H. WHO classification of the chronic myeloproliferative diseases (CMPD) polycythemia vera, chronic idiopathic myelofibrosis, essential thrombocythemia and CMPD unclassifiable. Tumours of Haematopoiesis and Lymphoid Tissues. Lyon. 2001; 31-42.
- Michiels JJ, De Raeve H, Berneman Z, Van Bockstaele D, Hebeda K, et al. The 2001 World Health Organization (WHO) and updated European clinical and pathological (ECP) criteria for the diagnosis, classification and staging of the Ph1-chromosome negative chronic myeloproliferative disorders (MPD). Sem Thromb Hemostas. 2006; 32: 307-340.
- Thiele J, Kvasnicka HM, Diehl V, Fischer R, Michiels JJ. Clinicopathological diagnosis and differential criteria of thrombocythemias in various myeloproliferative disorders by histopathology, histochemistry and immunostaining from bone marrow biopsies. Leukemia and Lymphoma. 1999; 33: 207-218. Ref.: http://tinyurl.com/yy79mqsk
- Thiele J, Kvasnicka HM. Chronic myeloproliferative disorders with thrombocythemia: a comparative study of two classifications systems (PVSG-WHO) on 839 patients. Ann Hematol. 2003; 82: 148-152. Ref.: http://tinyurl.com/y3hryz69
- Florena AM, Tripodo C, Iannitto E, Porcasi R, Ingrao S, et al. Value of bone marrow biopsy for diagnosis of essential thrombocythemia. Haematologica. 2004; 89: 911-919. Ref.: http://tinyurl.com/y4b9f3qd
- Lengfelder E, Hochhaus A, Kronawitter U. Should a platelet count of 600 x109/l be used as a diagnostic criterion in essential thrombocythemia? An analysis of the natural course including early stages. Br J Haematol. 1998; 100: 15-23.
- Sacchi S, Vinci G, Gugliotta L, Rupoli S, Garganti L, et al. Diagnosis of essential thrombocythemia at platelet counts between 400 and 600x109/l. Gruppo Italiano Malattie Mieloproliferative Chroniche (GIMMC). Haematologica. 2000; 85: 492-495. Ref.: http://tinyurl.com/y3setqtg
- Michiels JJ, Ten Kate FWJ. Erythromelalgia in thrombocythemia of various myeloproliferative disorders. Am J Hematol. 1992; 39: 131-136. Ref.: http://tinyurl.com/yxr8gy7m
- Michiels JJ, Juvonen E. Proposal for revised diagnostic criteria of essential thrombocythemia and polycythemia vera by the Thrombocythemia Vera Study Group. Semin Thromb Hemostas. 1997; 23: 339-347. Ref.: http://tinyurl.com/y6zb6dah
- Wasserman LR. Polycthemia vera, its course and treatment: relation to myeloid metaplasia and leukemia. Bull NY Acad Med. 1954; 30: 343-375. Ref.: http://tinyurl.com/y5448f94
- Wasserman LR, Berk PD, Berlin NI. Polycythemia vera and the myeloproliferative disorders. WB Saunders Philadelphia. 1995; ISBN 0-7216-4213-6.
- Michiels JJ, Barbui T, Fruchtman SM, Kutti J, Rain JD, et al. Diagnosis and treatment of polycythemia vera and possible future study designs of the PVSG. Leukemia Lymphoma. 2000; 36: 239-253. Ref.: http://tinyurl.com/y5tm3zbp
- Pearson TC, Wetherley-Mein G. The course and complications of idiopathic erythrocytosis. Clin Lab Haematol. 1979; 1: 189-196. Ref.: http://tinyurl.com/y5u7m9rd
- Najean Y, Triebel F, Dresch C. Pure erythrocytosis: reappraisal of a study of 51 patients. Am J Hematol. 1981; 10: 129-136. Ref.: http://tinyurl.com/yxtn5zx6
- Kurnick JE, Ward HP, Block MH. Bone marrow sections in the differential diagnosis of polycythemia. Arch Path. 1972; 94: 489-499. Ref.: http://tinyurl.com/y3m2mn8y
- Ellis JT, Silver RT, Coleman M, Geller SA. The bone marrow in polycythemia vera. Sem Hematol. 1975; 12: 433-444. Ref.: http://tinyurl.com/y5afjdyz
- Ellis JT, Peterson P. The bone marrow in polycythemia vera. Pathol Annu. 1979; 14: 383-403. Ref.: http://tinyurl.com/y3c6dvks
- Prchal JF, Axelrad AE. Bone marrow responses in polycythemia vera. N Eng J Med. 1974; 290: 1382. Ref.: http://tinyurl.com/y6axsstc
- Prchal JF, Axelrad A, Crookston JH. Erythroid colony formation in plasma culture from cells of peripheral blood in myeloproliferative disorders. Blood. 1974; 44: 912.
- Zanjani ED, Lutton JD, Hoffman R, Wasserman LR. Erythroid colony formation by polycythemia vera bone marrow in vitro. Dependence on erythropoietin. J Clin Invest. 1977; 59: 841-848. Ref.: http://tinyurl.com/y2xqsvpy
- Casadevall N, Lacombe C, Varet B. In vitro study of erythroid. Precursors in Vaquez’s disease (polycythemia vera). Evidence supporting 2 populations of elytroid. Stem cells in the bone marrow. Nouvelle Revue Francaise d’Hematologie. 1978; 20: 575-574. Ref. : http://tinyurl.com/yy3rfjc4
- Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol. 2002; 76: 133-145. Ref.: http://tinyurl.com/y3r2hp25
- Michiels JJ. Bone marrow histopathology and biological markers as specific clues to the differential diagnosis of essential thrombocythemia, polycythemia vera and prefibrotic or fibrotic myeloid metaplasia. Hematol J. 2004; 5: 93-102. Ref.: http://tinyurl.com/y2zlay6n
- Thiele J, Kvasnicka HM, Diehl V. Bone marrow features of diagnostic impact in erythrocytosis. Ann Haematol. 2005; 84: 362-367. Ref.: http://tinyurl.com/y47bqszj
- Thiele J, Kvasnicka HM, Diehl V. Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematologica. 2005; 113: 213-219. Ref.: http://tinyurl.com/y5ktlbk7
- Thiele J, Kvasnicka HM, Zankovich R, Diehl V. The value of bone marrow histopathology for the differentiation between early stage polycythemia vera and secondary (reactive) polycythemias. Haematologica. 2001; 86: 368-374. Ref.: http://tinyurl.com/y584phsq
- Thiele J, Kvasnicka HM, Muehlhausen K, Walter S, Zankovich R, et al. Polycythemia rubra vera versus secondary polycythemias. A clinicopathological evaluation of distinctive features in 199 patients. Pathology Res Pract. 2001; 197: 77-84. Ref.: http://tinyurl.com/y69jrm5p
- Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Blood. 2007. 110: 1092-1097. Ref.: http://tinyurl.com/y6kkf6ew
- Sirhan S, Fairbanks VG, Tefferi A. Red cell mass and plasma volume measurements in polycythemia. Cancer. 2005; 104: 213-215. Ref.: http://tinyurl.com/y642u8m9
- Johansson PL, Safia-Kutti S, Kutti J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: a study of patients with polycythemia vera and apparent polycythaemia. Br J Haematol. 2005; 129: 701-705. Ref.: http://tinyurl.com/y32628n5
- Westwood NB, Pearson TC. Diagnostic applications of haematopoietic progenitor culture techniques in polycythaemias and thrombocythaemias. Leukemia Lymphoma. 1996; 22: 95-103. Ref.: http://tinyurl.com/y4sdzk9l
- Juvonen E, Ikkala E, Oksanen K, Tapani R. Megakaryocyte and erythroid colony formation in essential thrombocythaemia and reactive thrombocytosis: diagnostic value and correlation to complication. Br J Haematol. 1993; 83: 192-197. Ref.: http://tinyurl.com/y6ez6b6k
- Shih LY, Lee CT. Identification of masked polycythemia vera from patients with idiopathic thrombocytosis by endogenous elytroid colony assay. Blood. 1994; 83: 744-748. Ref.: http://tinyurl.com/yxkfc9af
- Liu E, Jelinek J, Pastore YD, Guan Y, Prchal JF. Discrimination of polycythemias and thrombocytoses by novel simple, accurate clonality assays and comparison with PRV-1expression and BFU-e responses to erythropoietin. Blood. 2003; 101: 3294-3301. Ref.: http://tinyurl.com/y2he6wkm
- Dobo I, Donnard M, Giridon F, Mossuz P, Boiret N, et al. Standardization and comparison of endogenous erythroid colony assays performed with bone marrow or blood progenitors for the diagnosis of polycythemia vera. Hematol J. 2004; 5: 161-167. Ref.: http://tinyurl.com/y54dzoju
- Mossuz P, Giridon F, Latger-Cannard V, Dobo I, Boiret N, et al. Diagnostic value of serum erythropoietin level in patients with absolute erythrocytosis. Haematologica. 2004; 89: 1194-1198. Ref.: http://tinyurl.com/y32v63gk
- Johansson P, Andreason B, Safai-Kutti S, Wennstrom L, Palmqvist L, et al. The presence of a significant association between elevated PRV-1 mRNA expression and low plasma erythropoietin concentration in essential thrombocythemia. Eur J Haematol. 2003; 70: 358-362. Ref.: http://tinyurl.com/y2vxuu2z
- Temerinac S, Klippel S, Strunck E, Röder S, Lübbert M, et al. Cloning of PRV-1, a novel member of the uPAR receptor superfamily, which is over expressed in polycythemia rubra vera. Blood. 2000; 95: 2569-2576. Ref.: http://tinyurl.com/y2zrkrqp
- Pahl HL. Polycythaemia vera: will new markers help us answer old questions? Acta Haematol. 2002; 108: 120-131. Ref.: http://tinyurl.com/y5w3ccpo
- Goertler PS, Steimle C, Maerz E, Johanson PL, Andreasson B, Griesshammer M, et al. The JAK2 V617F mutation, PRV-1 over expression and EEC formation define a similar cohort of MPD patients. Blood. 2005; 106: 2862-2864. Ref.: http://tinyurl.com/y6bkgtal
- Griesshammer M, Klippel S, Strunk E, Temeric S, Mohr U, et al. PRV-1 mRNA expression discriminates two types of essential thrombocythemia. Ann Hematol. 2004; 83: 364-370. Ref.: http://tinyurl.com/y5el3guo
- Messinezy M, Westwood NB, El-Hemaida I, Marsden JT, Sherwood RS, et al. Serum erythropoietin values in erythrocytoses and in primary thrombocythaemia. Br J Haematol. 2002; 117: 47-53. Ref.: http://tinyurl.com/yytm66zv
- Jantunen R, Juvonen E, Ikkala E, Oksanen K, Antilla P, et al. Development of erythrocytosis in the course of essential thrombocythemia. Ann Hematol. 1999; 78: 219-222. Ref.: http://tinyurl.com/y5hljytl
- De Stefano V, Teofili L, Leone G, Michiels JJ. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. Sem Thromb Hemostas. 1997; 23: 411-418. Ref.: http://tinyurl.com/y6d2n38q
- Chait Y, Condat B, Cazals-Hatem D, Rufat P, Atmani S, et al. Relevance of the criteria commonly used to diagnose myeloproliferative disorders in patients with splanchnic vein thrombosis. Br J Haematol. 2005; 129: 553-560. Ref.: http://tinyurl.com/y552tgae
- Brière J. Budd-Chiari syndrome and portal vein thrombosis associated with myeloprioliferative disorders: diagnosis and management. Sem Thromb Hemostas. 2006; 32: 208-218. Ref.: http://tinyurl.com/yxaz3nfc
- Smalberg JH, Murad SD, Braakman E, Valk PJ, Janssen LA, et al. Myeloproliferative disease in the pathogenesis and survival of Budd-Chiari syndrome. Haematologica. 2006; 91: 1712-1713. Ref.: http://tinyurl.com/yyeelyr3
- Patel RK, Lea NC, Heneghan A, Westwood N, et al. Prevalence of the activating JAK2 troikas mutation V617F in the Budd-Chiari Syndrome. Gastroenterology. 2006; 130: 2031-2038. Ref.: http://tinyurl.com/y38kh7l5
- Colaizzo D, Amitrano L, Tiscia L, Scenna G, Grandone E, et al. The JAK2 V617F mutation frequently occurs in patients with portal vein and mesenteric vein thrombosis. J Thromb Haemostas. 2007; 5: 55-61. Ref.: http://tinyurl.com/y5pongum
- De Stefano V, Fiorini A, Rossi E, Farina G, Reddiconto G, et al. Prevalence of the JAK2V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J Thromb Haemostas. 2007; 4: 708-714. Ref.: http://tinyurl.com/y5emhuk5
- Pragmanini M, Barosi G, Berrgamaschi G, Fianelli U, Fabris F, et al. Role of the JAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology. 2006; 44: 1528-1534. Ref.: http://tinyurl.com/y4xbdctc
- Boissinot M, Lippert E, Girodon F, Dobo I, Fouassier M, et al. Latent myeloproliferative disorder revealed by the JAK2V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood. 2006; 108: 3323-3324. Ref.: http://tinyurl.com/y24xpvmt
- Vainchenker W, Delhommeau F, Villeval JL. Molecular pathogenesis of the myeloproliferative diseases. Hematology Education, EHA. 2007; 1: 239-246.
- Delhommeau F, Pisani DF, James C, Casadevall N, Constatinescu S, et al. Oncogenic mechanism in myeloproliferative disorders. Cell Mol Life Sci. 2006; 63: 2939-2953. Ref.: http://tinyurl.com/y2zaghxz
- Villeval JL, James C, Pisani DF, Casadevall N, Vainchenker W. New insights into the pathogenesis of JAK2V617F-positive myeloproliferative disorders and consequences for the management of patients. Sem Thromb Hemostas. 2006; 32: 341-351. Ref.: http://tinyurl.com/y5f64xjw
- Michiels JJ, Berneman Z, Van Bockstaele D, Van Der Planken M, De Raeve H, et al. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Sem Thromb Hemostas. 2006; 32: 174-207. Ref.: http://tinyurl.com/y6anppg8
- Passamonti F, Rumi E, Pietra D, Della Porta MG, Boveri E, et al. Relation between JAK2 V617F mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006; 107: 3676-3682. Ref.: http://tinyurl.com/yyxpyaxc
- Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F JAK2 mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006; 108: 2435-2437. Ref.: http://tinyurl.com/y2sa4gky
- Dupont S, Massé A, James C, et al. The JAK2V617F mutation triggers erythropoietin hypersensitivity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood. 2007. 110: 1013-1021. Ref.: http://tinyurl.com/y249xmgs
- Tefferi A, Lasho TL, Schwager SM, Strand JS, Elliott M, et al. The clinical phenotype of wild-type, heterozygous, and homozygous JAK2V617F in polycythemia. Cancer. 2006; 106: 631-635. Ref.: http://tinyurl.com/y5ov7wnn
- Chen Z, Notohamiprodjo M, Guan XX, Paietta E, Blackwell S, et al. Gain of 9p in the pathogenesis of polycythemia vera. Genes Chromosomes & Cancer. 1998; 22: 321-324. Ref.: http://tinyurl.com/y4jwbh5p
- Naifeld V, Montella L, Scalise A, Fruchtman S. Exploring polycythemia vera with fluorescence in situ hybridization: additional cryptic 9p is the most frequent abnormality detected. Br J Haematol. 2002; 119: 558-566. Ref.: http://tinyurl.com/y3uaz2lt
- Campbell PJ, Baxter EJ, Beer PhA, Scott LM, Bench AJ, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and the role in leukemic transformation. Blood. 2006; 18: 3548-3555. Ref.: http://tinyurl.com/y56udcd2
- Scott LM, Tong W, Levine RL, Scott MA, Beer PA, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. New Eng J Med. 2007; 356: 459-468. Ref.: http://tinyurl.com/yycdnksg
- Zhou W, Toombs CF, Zou T, Guo J, Robinson MO. Transgenic mice overexpression human c-mpl ligand exhibit chronic thrombocytosis and display enhanced recovery from 5-fluoruracil or antiplatelet serum treatment. Blood. 1997; 89: 1551-1559. Ref.: http://tinyurl.com/y2sc452s
- Pikman Y, Lee BH, Mercher Th, McDowell E, Ebert BL, et al. MPLW515L is a novel somatic activation mutation in myelofibrosis with myeloid metaplasia. PLOS Med. 2006; 3: e270. Ref.: http://tinyurl.com/yyby9jua
- Pardanani A, Levine RL, Lasho TL, Pikman Y, Mesa RA, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006; 108: 3472-3476. Ref.: http://tinyurl.com/yyn35ppv
- Michiels JJ, Abels J, Steketee J, vanVliet HHDM, Vuzevski VD. Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med. 1985; 102: 466-471. Ref.: http://tinyurl.com/y6luwa68
- Michiels JJ, Koudstaal P, Mulder AH, van Vliet HHDM. Transient neurologic and ocular manifestations in primary thombocythemia. Neurology. 1993; 43: 1107-1110. Ref.: http://tinyurl.com/y3s6c22s
- Michiels JJ, van Genderen PJJ, Lindemans J, van Vliet HHDM. Erythromelalgic, thrombotic and hemorrhagic manifestations in 50 cases of thrombocythemia. Leukemia & Lymphoma. 1996; 22: 47-56. Ref.: http://tinyurl.com/y68eq38g
- Michiels JJ, Commandeur S, Hoogenboom GJ, Wegman JJ, Scholten L, et al. JAK2V617F positive early stage myeloproliferative disease (essential thrombocythemia) as the cause of portal vein thrombosis in two middle-aged females: therapeutic implications in view of the literature. An Hematol. 2007; 86: 793-800. Ref.: http://tinyurl.com/y377uo55
- Schlemper RJ, van der Maas APC, Eikenboom JCJ. Familial essential thrombocythemia: clinical charateristics of 11 cases in one family. Ann Hematol. 1994; 68: 153-158. Ref.: http://tinyurl.com/y24a7qfh
- Wiestner A, Schlemper RJ, van der Maas APC, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythemia. Nat Genet. 1998; 18: 49-52. Ref.: http://tinyurl.com/y68ns5pz
- Kralovics R, Buser AS, Teo SS, Coers J, Tchelli A, et al. Comparison of molecular markers in a cohort of patients with chronic myeloproliferative disorders. Blood. 2003; 102: 1869-1871. Ref.: http://tinyurl.com/y4mj7esj
- Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes the receptor for thrombopoietin. Blood. 2004; 103: 4198-4200. Ref.: http://tinyurl.com/y2nlrob5
- Georgii A, Buhr T, Buesche G, Kreft A, Choritz H. Classification and staging of Ph-negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leukemia and Lymphoma. 1996; 22: 15-29. Ref.: http://tinyurl.com/y6jy8pf7
- Georgii A, Buesche G, Kreft A. The histopathology of chronic myeloproliferative diseases. Bailière’s Clin Haematol. 1998; 11:721-749. http://tinyurl.com/y6s3o9ay
- Thiele J, Kvasnicka HM, Werden C, Zankovich R, Diehl Fischer R. Idiopathic primary osteomyelofibrosis: A clinico-pathological study on 208 patients with special emphasis on evolution of disease features, differentiation from essential thrombocythemia and variables of prognostic impact. Leukemia and Lymphoma. 1996; 22: 303-317. Ref.: http://tinyurl.com/y3ats2fq
- Thiele J, Kvasnicka HM, Diehl V, Fischer R, Michiels JJ. Clinicopathological diagnosis and differential criteria of thrombocythemias in various myeloproliferative disorders by histopathology, histochemistry and immunostaining from bone marrow biopsies. Leukemia and Lymphoma. 1999; 33: 207-218. Ref.: http://tinyurl.com/yy79mqsk
- Thiele J. Kvasnicka HM, Fischer R. Histochemistry and morphometry on bone marrow biopsies in chronic myeloproliferative disorders: aids to diagnosis and classification. Ann Hematol. 1999; 78: 495-506. Ref.: http://tinyurl.com/y4y65hqm
- Thiele J, Kvasnicka HM. Clinicopathological criteria for the differential diagnosis of thrombocythemia in various myeloproliferative disorders. Sem Thromb Hemostas. 2006; 32: 219-230. Ref.: http://tinyurl.com/y63uwo4e
- Thiele J, Kvasnicka HM. A critical reappraisal of the WHO classification of the chronic myeloproliferative disorders. Leukemia and Lymphoma. 2006; 47: 381-396. Ref.: http://tinyurl.com/y55rx9bo
- Thiele J, Kvasnicka HM. Hematologic findings in chronic idiopathic myelofibrosis. Sem Oncol. 2005; 32: 380-304.
- Kvasnicka HM, Thiele J. The impact of clinicopathological studies on staging and survival in ET, PV and IMF. Sem Thromb Hemostas. 2006; 32: 362-371. Ref.: http://tinyurl.com/y56qnsts
- Barosi G, Ambrosetti A, Finelli C, et al. The Italian consensus on diagnostic criteria for myelofibrosis with myeloid metaplasia. Br J Haematol. 1999; 104: 730- 737. Ref.: http://tinyurl.com/y4rtft54
- Barosi G, Myelofibrosis with myeloid metaplasia: diagnostic definition and prognostic classification for clinical studies and treatment guidelines. J Clin Oncol. 199; 17: 2954-2970. Ref.: http://tinyurl.com/y2yjf2zo
- Michiels JJ, Kutti J, Stark P, Bazzan M, Gugliotta L, et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thromboythemia, polycythemia vera and essential megakaryocytic granulocytic myeloproliferation and myelofibrosis. Neth J Med. 1999; 54: 46-62. Ref.: http://tinyurl.com/y2czw485
- Michiels JJ, De Raeve H, Hebeda K, Lam KH, Berneman Z, et al. WHO bone marrow features and European clinical molecular and pathlogical criteria for the diagnosis and classification of myeloproliferative disorders. Leuk Res. 2007; 31: 1031-1038. Ref.: http://tinyurl.com/yyzdsylv
- Gianelli U, Vener C, Ravielle PR, Moro A, Savi F, et al. Essential thrombocythemia or chronic myelofibrosis? A single-center study based on hematopoietic bone marrow histology. Leuk Lymph. 2006; 47: 1774-1781. Ref.: http://tinyurl.com/yyw3m36e
- Kreft A, Buche G, Ghalibafian M, Buhr T, Fischer T, et al. The incidence of myelofibrosis in essential thrombocythemia, polycythemia vera and chronic idiopathic myelofibrosis: a retrospective evaluation of sequential bone marrow biopsies. Acta Haematol. 2005; 113: 137-143. Ref.: http://tinyurl.com/y5yhsnmh
- Thiele J, Kvasnicka HM, Schmitt-Graeff A, Zankovich R, Diehl V. Follow-up examinations including sequential bone marrow biopsies in essential thrombocythemia (ET): a retrospective clinicopathological study of 120 patients. Am J Hematol. 2002; 70: 283-291. Ref.: http://tinyurl.com/yye9q8z5
- Cervantes F, Alvarez-Larran A, Talarn C, Gomez M, Montserrat E. Myelofibrosis with myeloid metaplasia following essential thrombocythaemia: actuarial probability, presenting characteristics and evolution in a series of 195 patients. Br J Haematol. 2002; 118: 786-790. Ref.: http://tinyurl.com/y4q6enlj
- Wolansky A, Schwager SM, McClure RF, Larson DR, Tefferi A. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc. 2006; 81: 159-166. Ref.: http://tinyurl.com/yyfrdndl
- Michiels JJ, Berneman ZW, Schroyens W, Kutti J, Swolin B, et al. Philadelphia (Ph) chromosome positive thrombocythemia without features of chronic myeloid leukemia in peripheral blood: natural history and diagnostic differentiation from Ph-negative essential thrombocythemia. Ann Hematol. 2004; 83: 504-512. Ref.: http://tinyurl.com/yyvjp23w
- Schmitt-Graeff A, Thiele J, Zuk I, Kvasnicka HM. Essential thrombocythemia with ringed sideroblasts: a heterogenous spectrum of diseases, but not a distinct entity. Haematologica. 2002; 87: 392-399. Ref.: http://tinyurl.com/y4xcorpl
- Shaw GR. Ringed sideroblasts with thrombocytosis: an uncommon mixed myelodysplastic/myeloproliferative disease of older adults. Br J Haematol. 2005; 131: 180-184. Ref.: http://tinyurl.com/y2zev2we
- Szpurka H, Tiu R, Murugesan G, Aboudola S, His ED, et al. Refractory anemia with ringed sideroblasts associated with matked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2V617F mutation. Blood. 2006; 108: 2173-2181. Ref.: http://tinyurl.com/y2clq4e2
- Gatterman N, Billiet J, Kronenwett R, Zipperer E, Germing U, et al. High frequency of the JAK2 V617F mutation in patients with thrombocytosis (platelet count >600 x109/l) and ringed sideroblasts more than 15% considered as MDS/MPD, unclassifiable. Blood. 2007; 109: 1334-1335. Ref.: http://tinyurl.com/y24wxwvc
- Campbell P, Scott LM, Buck G, Wheatley K, East CL, et al. Definition of essential thrombocythemia and relation of essential thrombocythemia to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005; 366: 1945-1953. Ref.: http://tinyurl.com/y3xzxh56
- Campbell P, Green AR. The myeloproliferative disorders. New Eng J Med. 2006; 355: 2452-2466. Ref.: http://tinyurl.com/y4ymfgtq
- Juvonen E, Ikkala EE, Fyrrquist F Ruutu T. Autosomal dominant erythrocytosis caused by increased sensitivity to erythropoietin. Blood. 1991; 78: 3066-3069. Ref.: http://tinyurl.com/y45nwdoc
- De La Chapelle A, Träskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci. 1993; 90: 4495-4499. Ref.: http://tinyurl.com/y3fxnxua
- James C, Delhommeau F, Marzac C, Teyssandier I, Couédic JP, et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia 2006: 20: 350-353. Ref.: http://tinyurl.com/y4lpcrvp
- Tefferi A, Pardanani A. Mutation screening for JAK2V617F: when to order the test and how to interpret the results. Leukemia Res. 2006; 30: 739-744. Ref.: http://tinyurl.com/yylj489j
- Adamson JW. The erythropoietin/hematocrit relationship in normal and polycythemic man: implicatons of marrow regulation. Blood. 1968; 32: 597-609. Ref.: http://tinyurl.com/y2wxoxcb
- Napier JAF, Janowsky-Wieczorck A. Erythropoietin measurements in the differential diagnosis of polycythemia. Br J Haematol. 1981; 48: 393-401. Ref.: http://tinyurl.com/y3tdvk3e
- Cotes PM, Dore CJ, Tin JA, Lewis SM, Messinezy M, et al. Determination of serum immunoreactive erythropoietin in the investigation of erythrocytosis. N Eng J Med. 1986; 315: 283-287. Ref.: http://tinyurl.com/yyz5ruu2
- Birgegard G, Wide L. Serum erythropoietin in the diagnosis of polycythemia and after phlebotomy treatment. Br J Haematol. 1992; 81: 603-606. Ref.: http://tinyurl.com/yy8mgudn
- Messinezy M, Westwood NB, Woodstock SP, Strong RM, Pearson TC. Low serum erythropoietin: a strong diagnostic criterion of primary polycythaemia even at normal haemoglobin levels. Clin Lab Haematol. 1995; 17: 217-220. Ref.: http://tinyurl.com/y45p6syc
- Tefferi A. The diagnosis of polycythemia vera: new tests and old dictums. Best Practice & Research Clin Haematol. 2006; 19: 455-469. Ref.: http://tinyurl.com/y5v32o5b
- Andreasson B, Löfvenberg E, Westin J. Management of patients with polycythemia vera: results of a survey among Swedish haematologists. Eur J Haematol. 2005; 74: 489-495. Ref.: http://tinyurl.com/y5rwvlzb
- Le Bousse-Kerdiles MC, Martyré MC. Dual implication of fibrogenic cytokines in the pathogenesis of fibrosis and myeloproliferation in myeloid metaplasia with myelofibrosis. Ann Hematol. 1999; 78: 437-444. Ref.: http://tinyurl.com/y6fw95c2
- Bauermeister DE. Quantification of bone marrow reticulin. Am J Clin Pathol. 1971; 56: 24-31. Ref.: http://tinyurl.com/yyn6h6gc
- Manoharan A, Smart RC, Pitney WR. Prognostic factors in myelofibrosis. Pathology. 1982; 14: 445-461. Ref.: http://tinyurl.com/y3xrseho
- Thiele J, Kvasnicka HM, Facchetti F, Franco V, Van Der Walt J, et al. European consensus for grading of bone marrow fibrosis and assessment of cellularity in myeloproliferative disorders. Haematologica. 2005; 90: 1128-1132. Ref.: http://tinyurl.com/y59wzkm7
- Moliterno AR, Williams DM, Rogers O, Spivak JL. Molecular mimicry in the chronic myeloproliferative disorders: reciprocity between quantitative JAK2V617F and MPL expression. Blood. 2005; 106: 3520. Ref.: http://tinyurl.com/y6e5o6ec
- Bock O, Busche G, Koop C, Schroter S, Buhr T, et al. Detection of the single hotspot mutation in the JH2 pseudokinase domain of Janus kinase 2 in bone marrow trephine biopsies derived from myeloproliferative disorders. J Mol Diagn. 2006; 8: 170-177. Ref.: http://tinyurl.com/yydugqrw
- Bock O, Neuse J, Hussein K, Brakensiek K Buesche G, et al. Aberrant collagenase expression in chronic idiopathic myelofibrosis (CIMF) is related to the stage of disease but not to the JAK2 mutation status. Am J Pathol. 2006; 169: 471-481. Ref.: http://tinyurl.com/y26t9f9b
- Horn Th, Kremer M, Dechow T, Pfeifer WM, Geist B, et al. Detection of the activating JAK2 V617F mutation in paraffin-embedded trephine bone marrow biopsies of patients with chronic myeloproliferative diseases. J Mol Diagn. 2006; 3: 299-304. Ref.: http://tinyurl.com/y38u894j
- Michiels JJ, Kvasnicka HM, Thiele J. Myeloproliferative disorders. 2005 Verlag ME-Uwe Grunwald, Munich, Germany. ISBN. 3-9808075-6-8.
- Xu X, Zhang Q, Xing S, Li Q, Krantz A, et al. JAK2V617F: prevalence in a large Chinese hospital population. Blood. 2007; 109: 339-342. Ref.: http://tinyurl.com/y294tc8h
- Sidon EL Housni H, Dessars B, Heiman P. The JAK2V617F mutation is detectable at very low level in peripheral blood. Leukemia. 2006; 20: 1662. Ref.: http://tinyurl.com/yylj942p
- Passamonti F, Rumi E, Pietra D, Lazzarino M, Cazzola M. JAK2 (V617F) mutation in healthy individuals. Br J Haematol. 2007: 136: 677-679. Ref.: http://tinyurl.com/yxpguqjt
- Michiels JJ, De Raeve H, Berneman Z, Van Bockstaele D, Hebeda K, et al. The 2001 World Health Organization (WHO) and updated European clinical and pathological (ECP) criteria for the diagnosis, classification and staging of the Ph1-chromosome negative chronic myeloproliferative disorders (MPD). Sem Thromb Hemostas. 2006; 32: 307-340.
- Michiels JJ, Berneman Z, Van Bockstaele D, De Raeve H, Schroyens W. Current diagnostic criteria for the chronic myeloproliferative disorders (MPD) essential thrombocythemia (ET), polycythemia vera (PV) and chronic idiopathic myelofibrosis (CIMF). Pathologie Biologie. 2007; 55: 92-104. Ref.: http://tinyurl.com/yxrorx7s
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H. Changing concepts on the diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms. From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol. 2015; 133: 36-51. Ref.: http://tinyurl.com/y34d62ot
- De Raeve H, Fostier K, Valster F, Potters V, Kim Y, et al. Bone Marrow Histology is a Pathognomonic Clue to Each of the JAK2V617F, MPL515 and Calreticulin Mutated Thrombocythemia in Myeloproliferative Neoplasms. Clin Res Hematol 2018: 1: 1-7. Ref.: http://tinyurl.com/yycbcvnn
- De Raeve H, Michiels JJ, Valster F, Potters V, Kim Y, et al. Novel Clinical, Laboratory, Molecular and Pathological (2018 CLMP) Criteriafor the Differential Diagnosis of three Distinct JAK2, CALR and MPL MutatedMyeloproliferative Neoplasms: The Role of Driver Mutation Analysis and Bone Marrow Histology. Int J Cancer Res Ther. 2018; 3: 1-12.