
More Information
Submitted: 30 May 2019 | Approved: 20 June 2019 | Published: 21 June 2019
How to cite this article: Michiels JJ, Kim Y, Kim M, Valster F, Potters V, et al. Bone marrow histology in CALR mutated thrombocythemia and myelofibrosis: Results from two cross sectional studies in 70 newly diagnosed JAK2/MPL wild type thrombocythemia patients. Int J Bone Marrow Res. 2019; 2: 064-078.
DOI: 10.29328/journal.ijbmr.1001006
Copyright License: © 2019 Michiels JJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Myeloproliferative neoplasms; Essential thrombocythemia; Primary megakaryocytic granulocytic myeloproliferation; Myelofibrosis; Calreticulin mutation; JAK2 wild type; Bone marrow histology

Bone marrow histology in CALR mutated thrombocythemia and myelofibrosis: Results from two cross sectional studies in 70 newly diagnosed JAK2/MPL wild type thrombocythemia patients
Jan Jacques Michiels1,5*, Yonggoo Kim2,3, Myungshin Kim2,3, Francisca Valster4, Vincent Potters5, Zwi Berneman6, Alain Gadisseur6, Wilfried Schroyens6 and Hendrik De Raeve7
1Hematology, Coagulation and Vascular Medicine Research Center, Free University Network Europe, Goodheart Institute and Foundation in Nature Medicine & Health, Rotterdam, Netherlands
2Department of Laboratory Medicine, College of Medicine, The Catholic University of Korea, South Korea
3Catholic Genetic Laboratory Center, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, South Korea
4Department of Hematology, Bravis Hospital, Bergen op Zoom, Netherlands
5Department of Pathology, Bravis Hospital, Bergen op Zoom, Netherlands
6Department of Hematology, University Hospital Antwerp, Edegem, Belgium
7Departments of Pathology, OLV Hospital Aalst and University Hospital Brussels, Belgium
*Address for Correspondence: Dr. Jan Jacques Michiels, Professor, Goodheart Institute & Foundation in Nature Medicine, Freedom of Science & Education, Erasmus Tower, Veenmos 13, 3069 AT Rotterdam, Netherlands, Tel: +316-26970534; Email: [email protected]
The clinical phenotypes in 268 JAK2V617F mutated MPN patients in the Seoul study were PV in 101, ET in 95 and MF in 78 and 56 CALR mutated MPN consisted of PV in none, ET in 40 and MF in 16 cases. CALR mutated MPN patients were younger than JAK2V617F mutated MPN patients (mean ages 57.5 and 66 years), had lower values for values for leukocytes (8.6 vs 11.9x109/L) and higher values for platelets (898 vs 643x109/L respectively). Bone marrow histopathology in 268 JAK2V617F mutated MPN patients in the Seoul study was featured by an increased erythropoiesis and megakaryopoiesis (EM) in 13.5%, an increased erythropoiesis, megakaryopoiesis and granulopoiesis (EMG) in 31.3%, a normocellular megakaryocytic (M) proliferation in 29,1%, a megakaryocytic and granulocytic (MG) proliferation with a relative reduction of erythropoiesis in post-ET and Post-PV myelofibrosis in 26.2%. The bone marrow histology in 56 cases of CALR mutated MPN show a predominantly increased megakaryopoiesis (M) in two thirds and an increased megakaryopoiesis and granulopoiesis (MG) with a decreased erythropoiesis in one third.
Thirteen consecutive CALR MPN patients in the Belgian & Dutch cross sectional study presented with thrombocythemia associated with a typical PMGM bone marrow histology in 11 and myelofibrosis in 2 cases. All 11 thrombocythemia and 2 myelofibrosis CALR mutated MPN patients did not have constitutional symptoms and did not suffer from microvascular erythromelalgic disturbances, major thrombosis at platelet counts between 400 and 1000x109/L. There was an occurrence of hemorrhages at platelet counts above 1000x109/L in two CALR thrombocythemia cases.
Bone marrow histology of CALR mutated thrombocythemia in the Seoul and Belgian/Dutch study showed loose clusters of large megakaryocytes (M) with bulky, cloud-like nuclei with a normal or a minor reduction of erythropoiesis and no increase in reticulin fibers grade 0 or 1 (RF 0 or 1). CALR thrombocythemia patients show various degrees of increased bone marrow cellularity due to dual megakaryocytic and granulocytic (MG) proliferation featured by large megakaryocytes with roundish bulky nuclear forms and cloud-like clumsy nuclei, which are almost never seen in JAK2V617F ET and PV. Assessment of allele burden is an independent and most important factor for all molecular variants MPN disease burden. Overt myelofibrosis with advanced post PV and or ET myelofibrosis at the bone marrow level occurred in one third (30%) of 208 evaluable JAK2 MPN patients and in 8 (14%) of 56 CALR MPN patients in the Seoul study.
Polycythemia vera (PV) is a trilinear myeloproliferative disorder (MPD) of erythroid (E), megakaryocytic (M) and granulocytic (G) bone marrow proliferation first described by Dameshek (1950) and subsequently confirmed by the 1975 PVSG, 2001 WHO classifications of the myeloproliferative disorders (MPD) and the 2008 WHO classification of the myeloproliferative neoplasms (MPN) [1-7]. The minute Ph-chromosome in chronic myeloid leukemia (CML) originates from a translocation of chromosome 9 and 22 (t9:22), which results in a BRC/ABL fusion gene on chromosome 22 with high tyrosine kinase activity and CML- transforming capacity [8,9]. Michiels (1987) separated the Ph-chromosome BCR/ABL positive ET and CML from the Ph-chromosome BRC/ABL negative MPDs PV, ET or megakaryocytic leukemia (ML) without features of PV. The Ph+ / BRC/ABL positive ET and CML belong to one malignant disease with a rapidly progressive splenomegaly and myelofibrosis. The megakaryocytes are smaller than normal with mono- or binucleated nuclei in Ph+ / BCR/ABL positive ET and thrombocythemia associated with CML.
The PVSG defined primary myelofibrosis (PMF) is the third Ph-negative MPD entity without previous PV or ML or ET (Figure 1) [8]. In 1990 Georgii introduced the Hannover Bone Marrow classification of the Ph-negative MPD and revised the traditional 1975 PVSG and 1979 WHO classification of the MPDs by the introduction of bone marrow histology as a pathognomonic clue to each of MPD ET, PV and chronic primary megakaryocytic granulocytic myeloproliferation (CMGM/PMGM, (Figure 1) [8-26]. The BRC/ABL negative ET, PV and PMGM constitute the three distinct benign Ph-negative MPD entities ET, PV and CMGM (Figure 1). The characteristic increase in clustered medium to large-sized megakaryocytes and erythropoiesis with hyperplasia of dilatated sinuses are the diagnostic hallmark of untreated PV to distinguish it from secondary erythrocytosis and PMGM (Table 1) [8-26]. Bone marrow histology in PMGM is dominated by dysmorphic megakaryocytes, which are large due to increase in nuclear as well as cellular size [8-11]. The lightly stained chromatin and irregular roundish nuclear forms in PMGM are bulky with hyperlobulated and/or hypolobulated nuclei becoming clumpsy giving rise to the co-called cloud-like nuclei (Table 1), which are almost never seen in JAK2V617F and JAK2exon12 mutated ET and PV [8-11].
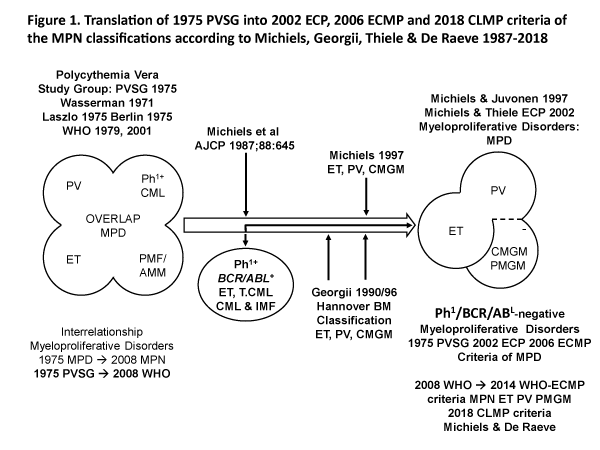
Figure 1: Classification of the myeloproliferative disorders (MPD) by the Polycthemia Vera Study Group (PVSG) in 1975 and WHO classifications in 1979 and 2001. Revision of the traditional PVSG classification into Ph+ ET and CML and the Ph- myeloproliferative disorders (MPDs) according to Michiels in 1987-1997 and Georgii (1990-1997) by the introduction of the Hannover Bone Marrow criteria as a pathognomonic clues to three distinct myeloproliferative disorders (MPD) essential thrombocythemia (ET), polycythemia vera (PV) and chronic or primary megakaryocytic granulocytic myeloproliferation (CMGM/PMGM) [1-6]. Blood and bone marrow features have been explicitly described in great detail by the ECP and ECMP classifications between 2005 and 2017 for the each of the JAK2V617F mutated ET and PV and MPL515 and for CALR mutated prefibrotic thrombocythemia and secondary myelofibrosis [1-26].
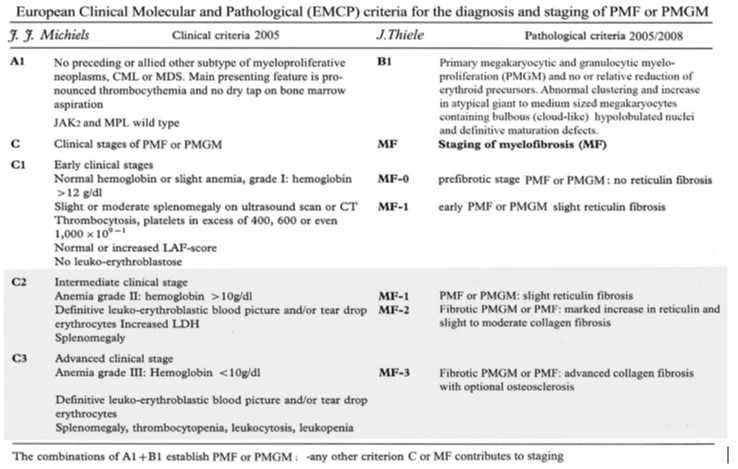
Table 1: European Clinical Molecular and Pathological (ECMP) criteria of JAK2/MPL wild type ET and MF carrying the CALR mutation: CALR-ET and CALR-MGM: Michiels & De Raeve 2005 – 2015.
The discovery in 2005 of the heterozygous and homozygous JAK2V617F mutation (Figure 1) as the driver cause of the trilinear MPNs in ET and PV patients by Constantinescu & Vainchenker [27,28] can easily explain the sequential occurrence of normocellular ET, prodromal PV, classical, masked and advanced PV complicated by secondary MF during lifelong follow-up. The JAK2V617F mutation load is low in heterozygous mutated ET, intermediate in heterozygous/homozygous prodromal PV and high in homozygous JAK2V617F mutated hypercellular and advanced PV [16-26]. Increase in large megakaryocytes with mature cytoplasm and multilobulated nuclei in a hypercellular bone marrow are more conspicuously altered in JAK2V617F mutated PV than in ET and prodromal stage PVm, MPL515 mutated ET is the second distinct MPN entity. The discovery of the calreticulin (CALR) somatic mutation in JAK2V617F/MPL515 wild ET and MF patients by Kralovics in 2013 [29], as the driver cause of ET associated with PMGM without features of PV (Figure 1, Table 1). CALR mutated hypercellular ET associated with PMGM has been recognized by Michiels & De Raeve in 2014 as the third distinct MPD entity without features of PV [20-26]. In this cross-sectional Belgian Dutch Korean (Seoul) collaborative study of 70 CALR mutated MPN cases we describe the clinical presentation, laboratory features and bone marrow characteristics in JAK2V617F mutated trilinear MPN and in CALR mutated thrombocythemia and myelofibrosis without features of PV.
Bone marrow histology methods
Bone marrow biopsies were performed from the iliac crest with an orthograde directed trephine which is a main prerequisite for bone marrow diagnosis of myeloproliferative neoplasms (MPNs) [14,15]. Fixation of the specimens is usually carried out in 4 % formalin. For achievement of optimal quality for enzyme- and or immunochemistry, any acid medium for decalcification has to be avoided. The next step in the present study consisted of paraffin embedding and employment of Giemsa, hematoxylin and eosin (H&E) staining, and silver impregnation method (Gomori’s techniques) [14,15]. Defining the early and advanced stages in each of the MPNs subtypes in routine practice include bone marrow cellularity related to various degrees of increased/decreased erythropoiesis and/or granulopoiesis, and more importantly the diagnostic differences of megakaryocyte number, size, morphology and clustering in the various clonally mutated MPNs ET, PV and PMGM [20-26]. The reticulin fiber content and pattern of fiber density in each patient at time of first diagnosis and in subsequent trephine biopsies is of utmost prognostic importance [9,10,14,15]. Bone marrow fibrosis (myelofibrosis = MF) is a secondary to the myeloproliferative transformation of bone marrow hematopoiesis in clonal JAK2V617F mutated ET and PV, MPL515 mutated ET and CALR ET / PMGM characteristics without features of PV (Figure 2) [9,30,31]. Myelofibrosis (MF) is according to Table 2 [14,15]. The blood and bone marrow features have been explicitly described in great detail by the ECP and ECMP classifications between 2005 and 2017 for the each of the JAK2V617F mutated ET and PV and MPL515 and for CALR mutated prefibrotic thrombocythemia and myelofibrosis (Table 2) [16-26]. Each of the MPDs JAK2V617F, MPL515 and CALR mutated ET, PV and PMGM as well as Ph+ET and Ph+CML are associated with various degrees of myeloid metaplasia of the spleen and secondary myelofibrosis [9,30,31].
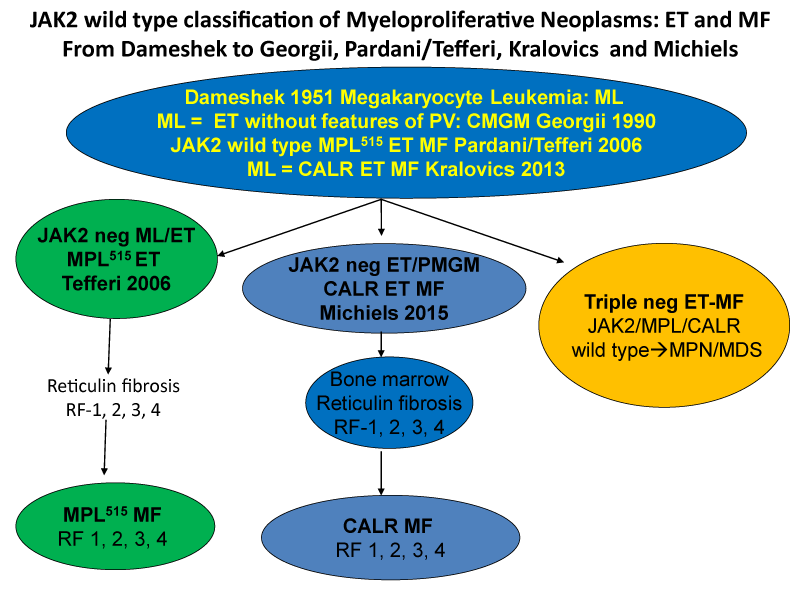
Figure 2:Classification of JAK2 wild type Myeloproliferative Neoplasms: ET and MF: From Dameshek (1951) to Georgii (1990), Pardani & Tefferi (2006) Kralovics (2013) and Michiels & De Raeve (2014, 2015, 2016, 2018) [23-26].

Table 2: Grading of myelofibrosis (MF) in myeloproliferative neoplasm (MPN): MF-0 Prefibrotic stage without or with an insignificant increase in reticulin content (RF grade 0 or +1); MF-1 Early fibrotic stage with a meshwork of reticulin fibers without collagen fibers (RF grade +2 ); MF-2 Fibrotic stage with a marked increase in reticulin with extensive intersections combined with collagen fibers (grade +3); MF-3 Advanced fibrotic stage with diffuse and dense increase in reticulin fibers RF with coarse bundles of collagen (RF grade +4) [14,15] .
Laboratory and bone marrow findings in JAK2 versus CALR MPN in the Seoul study
In the Seoul study of 407 MPN a driver mutation JAK2, CALR or MPL was detected in 82.7% with a mutation distribution of JAK2V617F in 275 (67.5%) and CALR in 55 (13.7%) and MPL in 6 (1.5% [30]). The clinical presentation and bone marrow features of the 6 MPL515 cases (ET in 3 and MF in 3) has been recently described by Michiels et al. 2018) [31]. The distribution of clinical phenotypes in 268 evaluable JAK2V617F mutated MPN in the Seoul study were PV in 101, ET in 95 and MF in 78 (Table 3). The distribution of clinical phenotypes in 56 CALR mutated MPN were PV in none, ET in 40 and MF in 16 cases (Table 3). The mean age of 56 CALR mutated MPN patients (57.5 years) was 8.5 years younger than in JAK2V617F mutated MPN patients (66 years). JAK2V617F mutated MPN had significantly higher values for leukocytes (11.9 x109/L) compared to CALR MPN (8.6x109/L) and lower values for platelets (643x109/L) compared to CALR MPN (898x109/L) (Table 3). Major thrombosis was recorded in 23 (7%) JAK2V617F mutated cases and in none of CALR mutated MPN (Table 3). The values for hemoglobin, hematocrit and erythrocyte counts in JAK2V617F mutated MPN ranged from anemic values in MF, to normal values in ET and increased values in PV (Table 3). CALR mutated MPN patients presented with decreased to normal values for hemoglobin, hematocrit and erythrocyte counts did not exceed the upper limit of normal.
| Table 3: Comparative analysis of the Clinical, Laboratory, Molecular and Pathologic (CLMP) characteristics of 324 WHO defined patients with Myeloproliferative Neoplasms (MPN) according to the driver mutations in 268 cases with the JAK2V617F and 56 cases with CALR genes in the cross-sectional Seoul MPN study of Kim et al. 2016 [30]. | ||
| 324 MPN patients | JAK2V617F | CALR |
| Patients N | 268 | 56 |
| Age yrs | 66 (22-89) | 56 (20-89) |
| Males (%) | 45.5% | 41.1% |
| Hemoglobin g/dL (range) | 14.7 (6.2-22.6) | 12.6 (7.5-16.1) |
| Hematocrit (%) (range) | 43.9 (19.7-69-1) | 38.4 (22.9-47.0) |
| Red Blood cells 1012/L (range) | 5.0 (1.89-9.92) | 4.2 (2.25-5.32 |
| Platelets 109/L (range) | 650 (13-3268) | 898 (49-1795) |
| Leukocytes 109/L (range) | 12.0 (2.2-177) | 8.6 (4.8-31) |
| 2008 WHO Class (N) | JAK2V617F | CALR |
| PV WHO | 101 | 0 |
| ET WHO | 95 | 40 |
| PMF WHO | 78 | 16 |
| BM CLMP Class (N) | JAK2 V617F | CALR |
| EM BM | 32 PV | 0 |
| EGM BM | 84 PV | 0 |
| M ET | 80 ET normocellular | 37 ET normocellular |
| MF 2/3 | 72 post ET PV MF | 19 MF |
| Mutation burden MB | MB JAK2 V617F | MB CALR |
| Total-Range | 67% (1.8-99) | 48% (17.8-93) |
| EM + EGM (PV) | 85% (13-99) | 0 |
| M (ET) | 37% ET normocell | *Type 1 53% |
| GM (MF) | 69% post ET PV MF | *Type 2 38% |
| Abbreviations: PV = polycythemia vera. ET = essential thrombocythemia. PMF = primary myelofibrosis. E = erythroid, M = megakaryocytic, G = granulocytic lineage myeloproliferation. N = number. Red = JAK2V617F mutated PV, ET or MF. Black = CALR mutated. *CALR type 1 revealed a higher mutant allele burden (53%) compared to CALR type 2 (38%) MPN. JAK2V617F mutated ET, ‘forme fruste’ prodromal PV and early PV patients who presented with a M or EM myeloproliferation showed no or minor (MF1/2) fibrosis. | ||
The bone marrow lineage proliferation profile in 268 evaluable cases of JAK2V617F mutated MPN featured dual increased proliferation of erythropoiesis and megakaryopoiesis (EM) in 13.5%, trilinear increased proliferation of erythropoiesis, megakaryopoiesis and granulopoiesis (EMG) in 31.3%, monolinear megakaryocytic proliferation (M) consistent with WHO define ET in 29,1% and dual megakaryocytic and granulocytic (MG) advanced hypercellular MPN with relative reduction of erythropoiesis mimicking PMGM in 26.2% (Table 3) [30]. The erythropoiesis was normal, relatively decreased or significantly decreased in prefibrotic and fibrotic stages of CALR mutated MPN (Table 3). The histology in 56 cases of CALR mutated MPN revealed a predominant increase in megakaryopoiesis (M) in a normal cellular bone marrow consistent with ET in two thirds and a dual increase in megakaryopoiesis and granulopoiesis (MG) consistent with PMGM in one third (Table 3).
The Clinical, Laboratory, Molecular and Pathologic (CLMP) characteristics of 268 evaluable JAK2V617F mutated patients and 56 CALR mutated patients in the cross sectional Seoul MPN study of Kim et al. 2016 [30], revealed that the frequency of myelofibrosis grade MF2/3 in 285 JAK2V617F was associated with an EMGM bone marrow in 22.2% and with a GM bone in 27.1% in 56 CALR-mutated PMGM patients. (Table 3). Myelofibrosis MF2/3 was present in 46% of advanced JAK2V617F+ GM and in 42% of advanced CALR-GM patients. Myelofibrosis MF2/3 was much lower in early stage JAK2V617F-M (17.5%) in JAK2V617F-EM (10.4%) patients and in early stage CALR-M (17.2%) (P<0.001)). None of the cases with initial stages of JAK2V617F -M normocellular ET or early stage prodromal PV patients with M or EM histology presented with MF2/3 grade fibrosis in the bone marrow (Table 3).
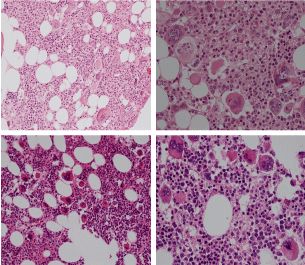
Figure 3: Bone marrow histopathology features for the distinction between large to giant mature megakaryocytes with hyperlobulated staghorn like nuclei in MPL515 mutated Thrombocythemia in case 10 (upper panels), Table 4 and large immature megakaryocytes with irregular roundish nuclear forms lobuli becoming clumsy give rise to the co-called cloud-like nuclei in CALR mutated Thrombocythemia in two asymptomatic cases of ET case 11 and 12 (lower panels), Table 4 with persistent increased platelet counts above as the only clinical presentation.
The correlations of bone marrow histology, grading of myelofibrosis, WHO clinical diagnosis and allele burden in 208 evaluable JAK2V617F mutated MPN patients of the Seoul study, table 4 shows that the majority of 57 JAK2 M patients have no or minimal fibrosis and allele burden, all 21 JAK2 EM PV patients have minimal fibrosis and high allele burden, 67 JAK2 EMG patients had minimal fibrosis and a high allele burden and about half of 63 JAK2 post ET/PV MF patients had an overt myelofibrosis at increased allele burden. These data in table 4 indicate that the assessment of the allele burden is an independent and important factor for MPN disease burden, whereas overt myelofibrosis at the bone marrow level occurred in about 30% of 208 evaluable JAK2 MPN patients, which is consistent with advanced post ET/PV myelofibrosis in the Seoul study.
| Table 4: Correlations of bone marrow histology, silver impregnation stained grading of myelofibrosis, WHO clinical diagnosis and allele burden in 208 evaluable JAK2V617F mutated MPN and 56 CALR mutated MPN patients in the Seoul study. | ||||||
| Bone Marrow Histology | JAK2 M | JAK2 EM | JAK2 EMG | JAK2 GM | CALR M | CALR MG |
| WHO Clinical Diagnosis | ET | PV | PV | ET/PV MF | ET | |
| Grading Myelofibrosis N | 57 | 21 | 67 | 63 | 39 | 19 |
| RF 0/1/2 MF 0/1 | 47 (83%) | 21 (110%) | 60 (90%) | 34 (54%) | 83% | 58% |
| Mutaton allele burden (%) | 33 | 74 | 89 | 58 | . | . |
| RF 3/4 MF 2/3 | 10 (17%) | 0 (0%) | 7 (10%) | 29 (46%) | 17% | 42% |
| Mutation allele burden (%) | 72 | . | 90 | 85 | ||
Results bone marrow histology of CALR thrombocythemia in the Seoul study
Bone marrow histology of WHO defined normocellular ET patients with typical prefibrotic CALR normocellular ET (CALR thrombocythemia) are featured by loose to dense clusters of large megakaryocytes with hypolobulated or hyperlobulated cloud-like nuclei, normal erythropoiesis and no increase in reticulin fibers RF grade 0. Bone marrow histology of four cases of CALR mutated normocellular thrombocythemia (ET) are shown in figures 4 and 5. A fifth case of CALR thrombocythemia diagnosed as ET showed a typical PMGM hypercellular bone marrow due to dual megakaryocytic granulocytic myeloproliferation with the presence of isolated large megakaryocytes, reduced erythropoiesis and reticulin fibrosis RF grade 1 (Figure 6 upper panels). A case of CALR mutated myelofibrosis (MF) showed a typical PMGM bone marrow featured by dense clusters of dysmorphic large megakaryocytes and cloud-like nuclei, reduced erythropoiesis and increase in reticulin fibers RF grade 2/3 (Figure 6 lower panels, Figure 7 upper panel). Another case of CALR mutated advanced myelofibrosis (MF) showed a myelofibrotic bone marrow with clustered large to giant immature megakaryocytes with cloud-like hypolobulated nuclei, reduced erythropoiesis and increase in reticulin RF grade 3 and presence of collagen bundles (Figure 7 lower panels).
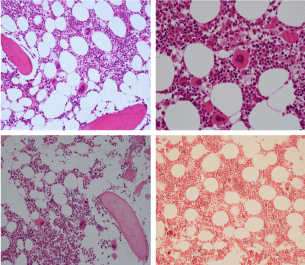
Figure 4: Two cases of CALR thrombocythemia in the Seoul study diagnosed as normocellular ET featured by the presence of large megakaryocytes with hypolobulated or hyperlobulated cloud-like nuclei, normal erythropoiesis and no increase in reticulin fibers (RF grade 0).
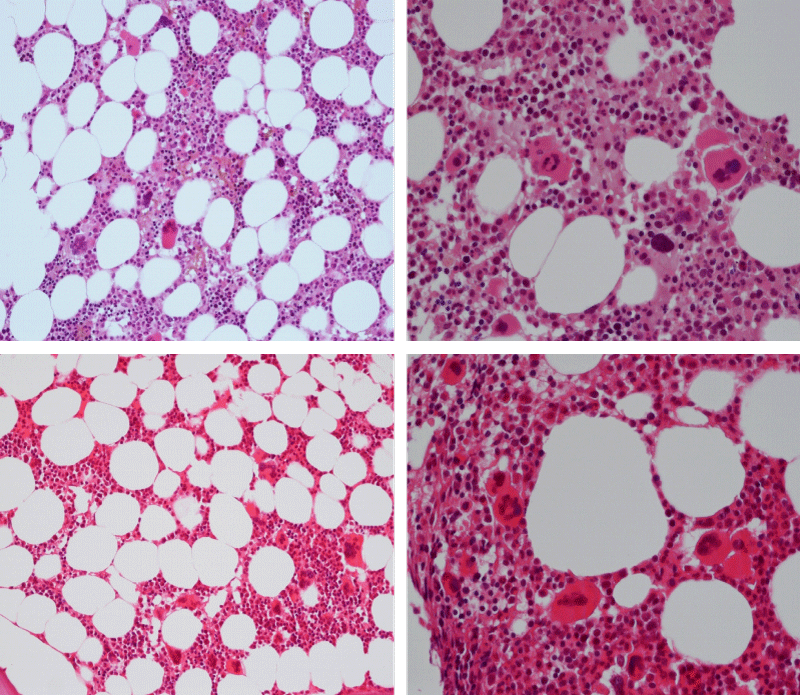
Figure 5: Two cases of CALR thrombocythemia in the Seoul study diagnosed as normocellular ET with the presence of loose clusters of large megakaryocytes with hypolobulated or hyperlobulated cloud-like nuclei, normal erythropoiesis and no increase in reticulin fibers (RF grade 0).
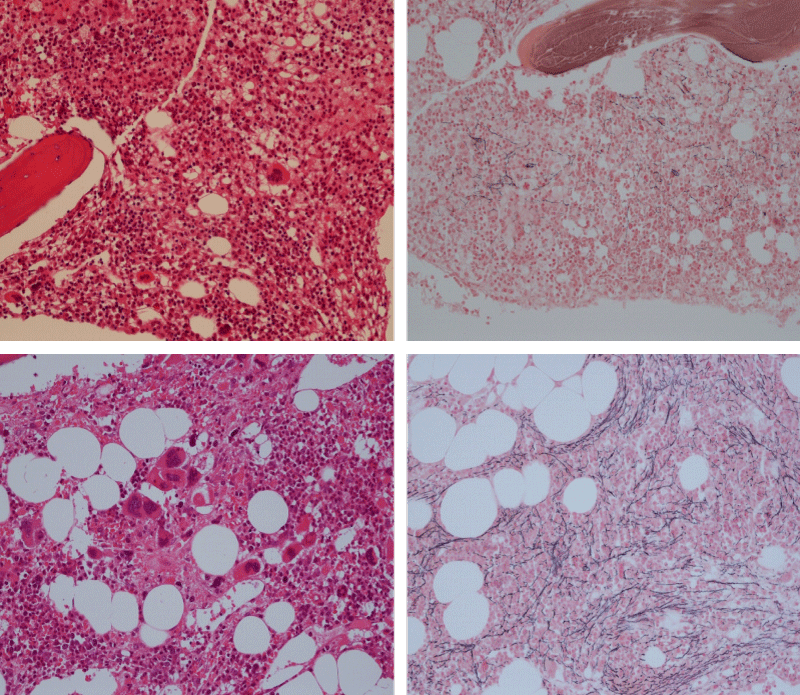
Figure 6: Upper panels. Case of CALR advanced myelofibrosis (MF) with dense clusters of large to giant megakaryocytes and cloud-like nuclei, reduced erythropoiesis and increase in reticulin fibers (RF grade 3). Lower panels. Case of CALR advanced myelofibrosis (MF) with clustered large to giant megakaryocytes with hypolobolated nuclei, reduced erythropoiesis and increase in reticulin and presence of collagen bundles (not stained in this silver impregnation technique) (RF grade 3).
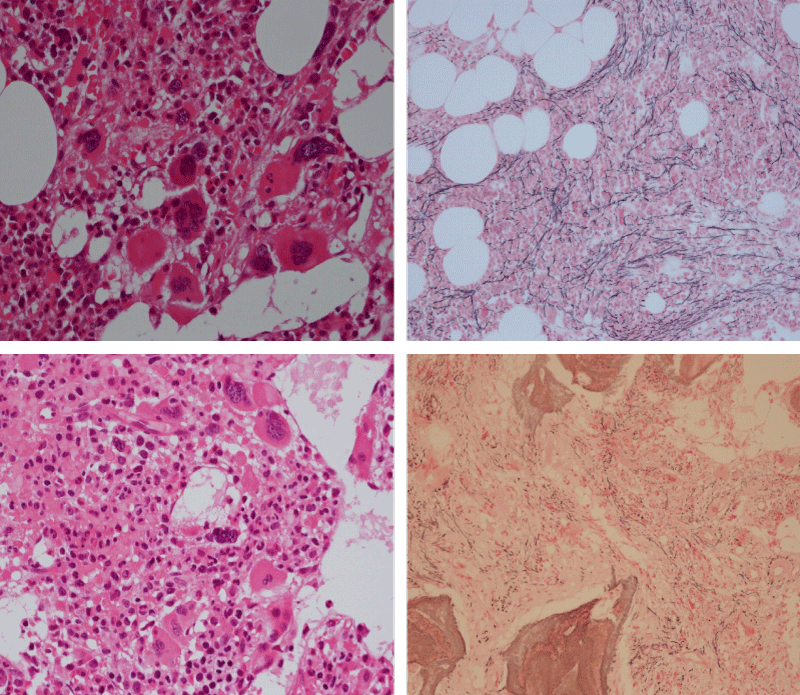
Figure 7: Upper panels. Case of CALR advanced myelofibrosis (MF) with dense clusters of large to giant megakaryocytes and cloud-like nuclei, reduced erythropoiesis and increase in reticulin fibers (RF grade 3). Lower panels. Case of CALR advanced myelofibrosis (MF) with clustered large to giant megakaryocytes with hypolobolated nuclei, reduced erythropoiesis and increase in reticulin and presence of collagen bundles (not stained in this silver impregnation technique) (RF grade 3).
Results of CALR thrombocythemia in the Belgian-Dutch study
Clinical and laboratory findings. Our series of 14 Belgian-Dutch consecutive JAK2 wild type MPN cases were recruited from the Antwerp region Flandria Belgium and South West Netherlands and diagnosed as CALR thrombocythemia in 13 and MPL thrombocythemia in one (Table 5). The CALR mutated thrombocythemia patients presented with prefibrotic ET associated with a typical PMGM bone marrow histology (ET/PMGM table 5) in 11 cases (85%). Two MF/PMGM cases (15%) (Case 5 and case 9, Table 4) presented with fatigue only. All 11 ET/PMGM and the 2 MF/PMGM were relative asymptomatic and not suffering from constitutional symptoms (Table 5). Two ET/PMGM cases presented with hemorrhagic manifestation at platelet count above 1000x109/L (Table 5). Case 2 presented with easy bruising at the age of 24 associated with documented acquired von Willebrand syndrome. Case 13 presented with subdural hematoma at platelet count of 1148x109/L, which was successfully treated by operative drainage and reduction of platelet count by hydroxyurea. The hemorrhagic thrombocythemia in these two CALR thrombocythemia patients was not preceded or followed by erythromelalgic microvascular circulation disturbances. Platelet counts in ET/PMGM ranged from 536 to 1306x109/L at time of first presentation and the two MF/PMGM cases had platelet counts of 265 and 347x109/L respectively. The values for hemoglobin, erythrocytes and white blood cells were in the normal range before and after follow-up in all cases of ET/PMGM (Table 1). Case 5 presented with advanced MF complicated by anemia and splenomegaly.
| Table 5: Clinical presentation, laboratory features, and pathological (CLMP) bone marrow (BM) histology diagnosis of 14 consecutive MPN cases with CALR mutated Thrombocythemia (ET) and primary megakaryocytic granulocytic BM proliferation (CALR PMGM) in eight CALR Thrombocythemia (ET) and megakaryocytic normocellular (CALR ET) bone marrow in three (case 6, 11 and 14), with clinical presentation of CALR MF in two (case 5 and 9) as compared to MPL515 Thrombocythemia (ET) and megakaryocytic (MPL M) BM proliferation in a normocellular bone marrow in case 10. Observations by Dr. Michiels and Dr De Raeve 2013-2018 (Figures 3 to 16). | ||||||||||
| Case/ | Year/ | HB mol/L | RBC | WBC | Platelets | LDH | Spleen | Presenting | Bone Marrow | BM histology |
| Gender | Age | g/dL/ Ht | x1012/L | x109/L | x109/L | U/L | size cm | symptoms | classification | MF/RF grading |
| 1 F | 2004 34 | 7.8 0.37 | 4.2 | 18.3 | 1230 | 1369 | 13 | Fatigue | CALR ET/MG | MF 0 RF 1 |
| 2 M | 2013 24 | 10.1 0.47 | 5.0 | 9.7 | 1306 | 390 | 16 | HT | CALR ET/MG | MF 0 RF 0 |
| 3 M | 2001 40 | 8.0 0.37 | 4.4 | 7.8 | 852 | N <620 | 13 | no | CALR ET/MG | MF 0 RF 0 |
| 2014 50 | 7.6 0.36 | 4.3 | 7.4 | 536 | N <620 | 16 | no | CALR ET/MG | MF 1 RF 2 | |
| 4 M | 2015 66 | 7.4 0.37 | 4.2 | 22.7 | 781 | nt | 20 | no | CALR ET/MG | MF 0 RF 1 |
| 5 M | 2015 68 | 5.2 0.26 | 3.3 | 5.9 | 265 | 452 | 24 | anemia | CALR MF | MF 3 RF 4 |
| 6 F | 2012 63 | 7.3 0.33 | 3.8 | 4.3 | 768 | 556 | 11.3 | no | CALR ET/M | MF 0 RF 0 |
| 7 F | 2014 64 | 7.5 0.40 | 4.3 | 6.1 | 1039 | 475 | 12 | no | CALR ET/MG | MF 0 RF 0 |
| 8 M | 2012 45 | 8.6 0.48 | 5.2 | 6.3 | 707 | 568 | 13 | no | CALR ET/MG | MF 0 RF 1 |
| 9 M | 2014 73 | 7.0 0.34 | 4.1 | 9.2 | 347 | 1519 | 14 | Fatigue | CALR MF | MF 2 RF 3 |
| 10 F | 2017 76 | 7.8 0.39 | 4.2 | 6.9 | 1243 | 475 | 12 | no | MPL515 ET/M | MF 0 RF 1 |
| 11 F | 2014 63 | 7.2 0.40 | 4.4 | 6.4 | 721 | 690 | np | no | CALR ET/M | MF 0 RF 1 |
| 12 M | 2011 74 | 13.6 0.41 | 4.3 | 11.4 | 714 | 785 | np | no | CALR ET/MG | MF 0 RF 1 |
| 13 M | 2014 75 | 11.5 0.35 | 4.0 | 5.8 | 1148 | 541 | np | HT | CALR ET/MG | MF 1 RF 2 |
| 14 F | 2015 58 | 14.4 0.42 | 5.2 | 6.6 | 965 | 530 | np | no | CALR ET/M | MF 0 RF 0 |
| CLMP = clinical, laboratory, molecular, and bone marrow classifification of CALR mutated MPN HT = hemorrhagic thrombocythemia and acquired von Willebrand syndrome type 2. The majority of CALR thrombocythemia were asymptomatic in terms of no constitutional symptoms and hemoglobin values in the normal range 7.2 to 10.1 mmol/L (in black) or increased 11.5 to 14.4 g/dL (in red). |
||||||||||
Bone Marrow Histology. Three cases of CALR mutated thrombocythemia (ET/PMGM case 3, 6, and 14) showed loose clusters of large megakaryocytes with bulky, cloud-like nuclei with a relative reduction of erythropoiesis and no increase in reticulin fibers grade 0 (RF 0) in a normocellular bone marrow with an abnormally increased megakaryopoiesis (M) indicative for MPN (Figures 8, 9). Two cases of CALR mutated thrombocythemia (ET/PMGM case 11 and 12, Figure 3) showed loose clustered dysmorphic megakaryocytes with the presence of fine reticulin fibers grade 1 (RF 1) in a normocellular M bone marrow in one (case 11) and in a hypercellular MG bone marrow in the other (case 12). Case 8 of CALR mutated ET/PMGM showed moderately dense clusters of medium-sized to large megakaryocytes with partly cloud-like nuclei with an area without reticulin fibers (RF 0) in a normocellular M bone marrow and an area with an increase in reticulin fibers (RF 2) in the hypercellular MG bone marrow area (Figure 11). Two cases of CALR mutated thrombocythemia (ET/PMGM case 3 after 12 years follow-up and case 9) showed an increase in reticulin fibers grade 2 (RF 2, Figures 12-14) and clusters of large megakaryocytes with dysmature, bulky, cloud-like nuclei in a slightly increased cellular bone marrow in case 9 and in the follow-up hypercellular MG bone marrow in case 3 (Figure 15). One case of CALR MF (MF/PMGM case 13) showed dense clustered large dysmorphic megakaryocytes with hyperlobulated nuclei in a hypercellular MG bone marrow with increased reticulin fibers (RF 2, Figure 14). The second case of CALR mutated MF (MF/PMGM case 5) presented with asymptomatic splenomegaly, no constitutional symptoms and symptomatic anemia associated with advanced myelofibrosis (RF 4, MF 3, Figure 16) in a hypocellular bone marrow showing dysmorphic megakaryocytes in fibrotic parts of the bone marrow. The transfusion dependent anemia responded to EPO, which improved the anemia from a Hb 4.2 Hb 5,7 mmol/L without the need of blood transfusion during follow-up.
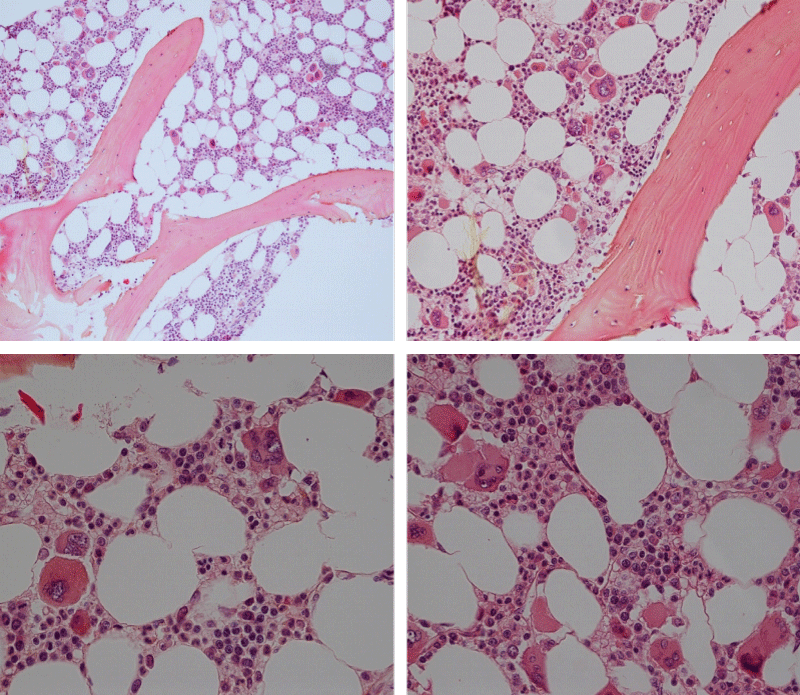
Figure 8: Bone marrow histology of CALR mutated ET/PMGM case 14 (female age 58) showing loosely clusters of large megakaryocytes with sometimes bulky, cloud-like nuclei in a completely normocellular bone marrow and no increase in reticulin fibers (RF grade 0/1).
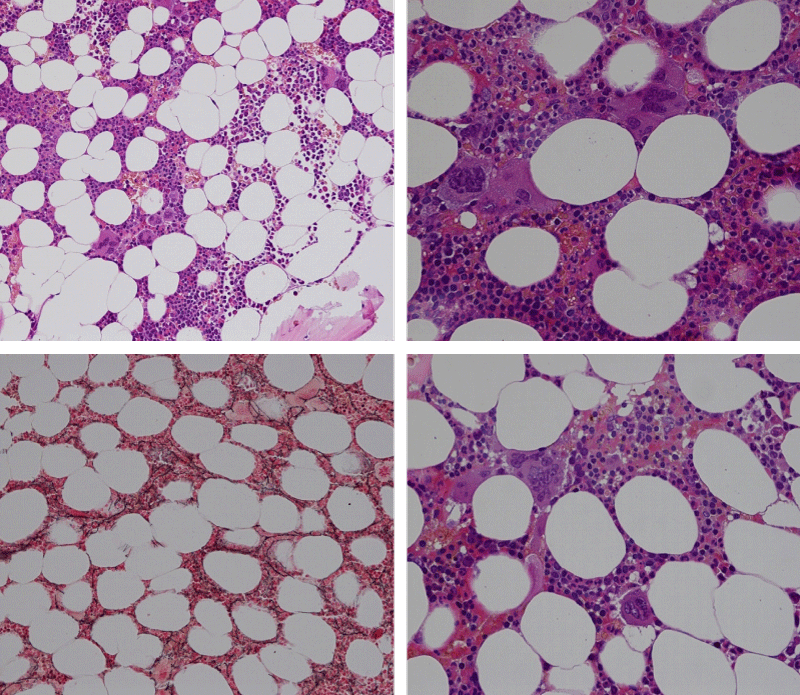
Figure 9: Bone marrow histology of CALR mutated ET/PMGM case 6 (female, age 63) showing moderately dense clusters of medium-sized and large megakaryocytes with sometimes dysmature cloud-like nuclei in a normocellular MGM bone marrow with a relative reduction of erythropoiesis and no distinct increase in reticulin fibers (RF grade 0/1).
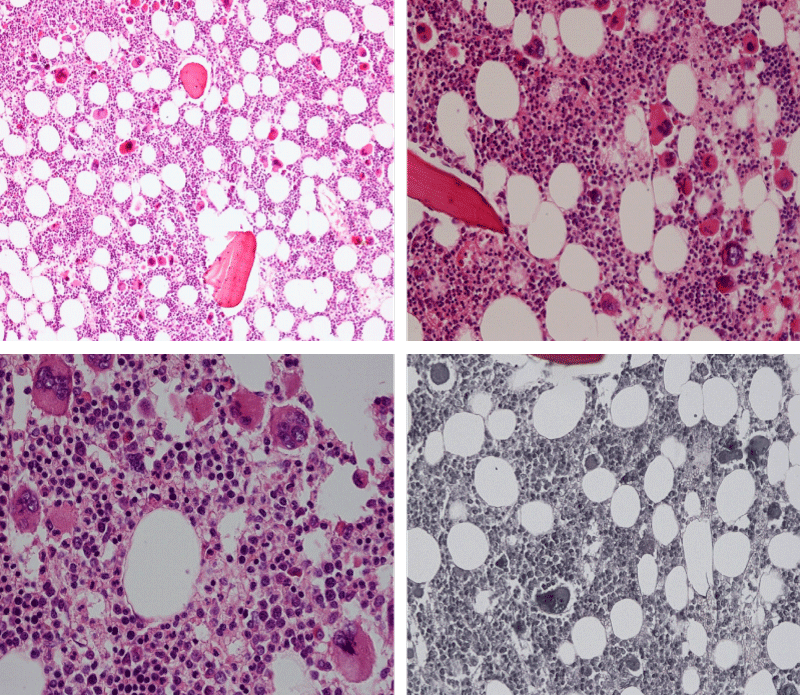
Figure 10: Bone marrow histology of CALR mutated ET/PMGM case 3 (man, age 39) showing dense clusters of large megakaryocytes with immature bulky, cloud-like nuclei in a hypercellular MGM bone marrow with a relative reduction of erythropoiesis and no increase in reticulin fibers (RF grade 0).
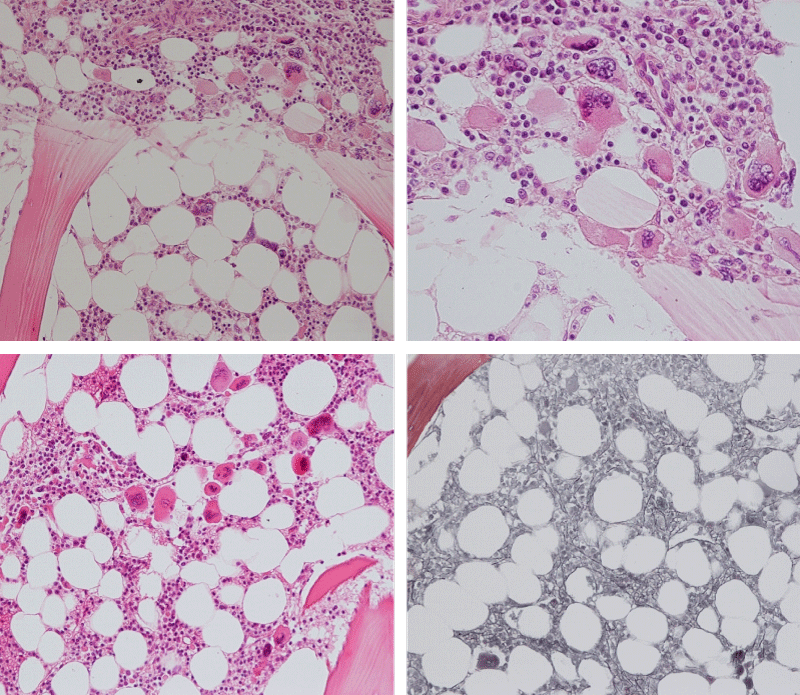
Figure 11: Bone marrow histology of CALR mutated ET/PMGM case 8 (man, age 39) showing moderately dense clusters of medium-sized to large megakaryocytes with partly cloud-like nuclei in a partly normocellular and partly hypercellular MGM bone marrow with no increase in reticulin fibers (RF grade 0) in normocellular area and with increase in reticulin fibers (RF grade 2) in hypercellular area of the bone marrow respectively.
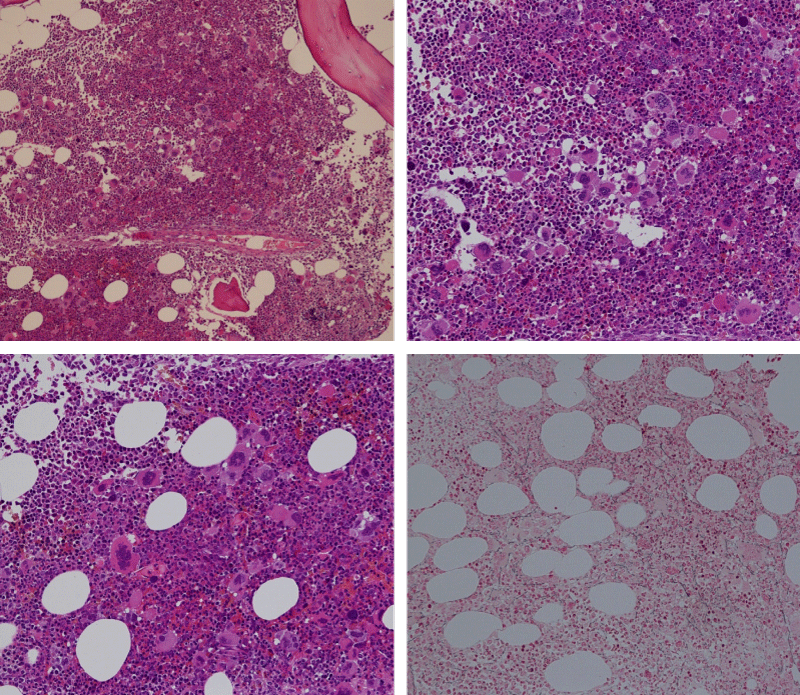
Figure 12: Hypercellular bone marrow due to a primary megakaryocytic granulocytic myeloproliferation (PMGM) in case 11 with CALR mutated thrombocythemia and reticulin fibers (RF grade 1).
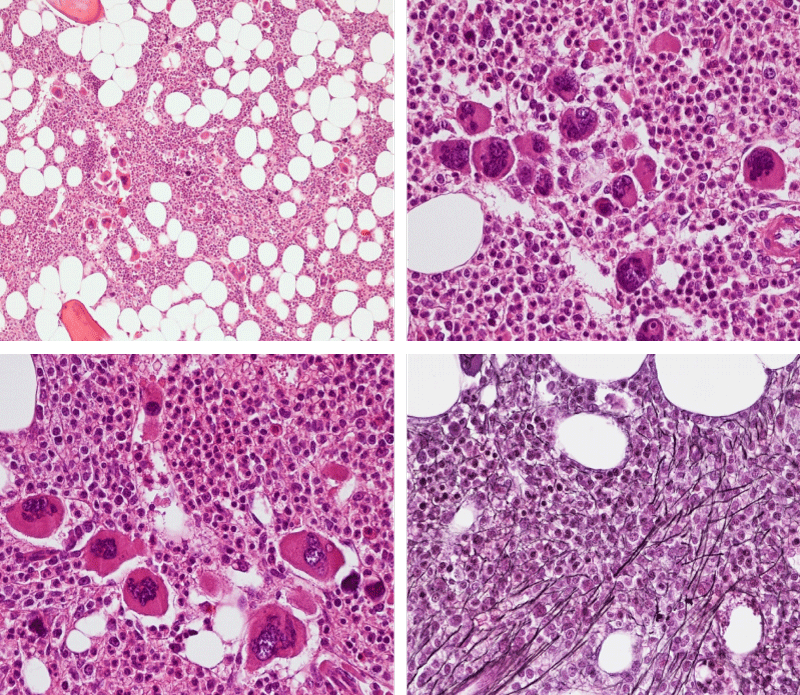
Figure 13: Bone marrow histology of CALR mutated ET/PMGM case 13 (female 58 years) showing dense clustered large dysmature megakaryocytes with hyperlobulated nuclei in a hypercellular bone marrow with increased reticulin fibers (RF grade 2).
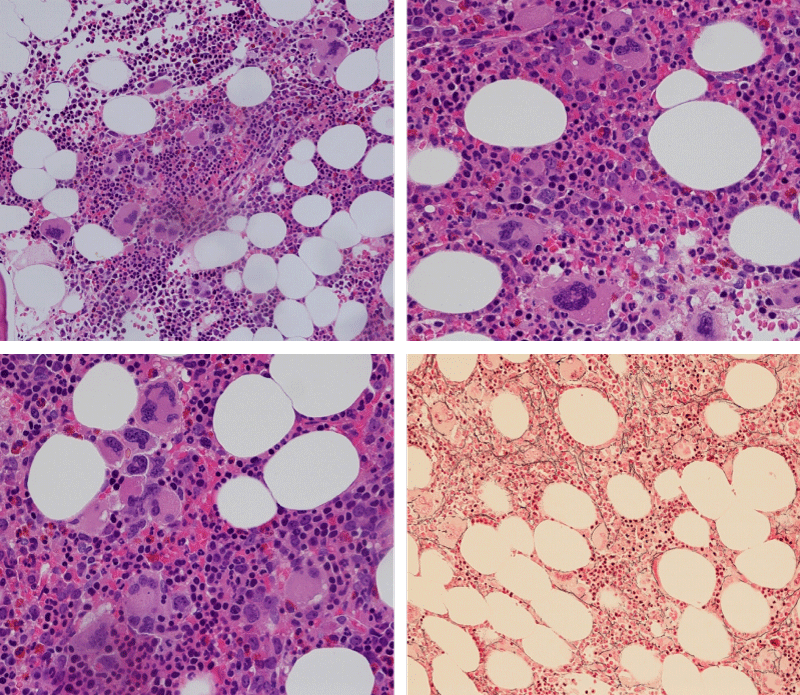
Figure 14: Bone marrow histology of CALR mutated MF/PMGM case 9 (man, age 73) showing loosely clusters of large megakaryocytes with dysmorphic, bulky, cloud-like nuclei in a slightly increased cellular bone marrow with an increase in reticulin fibers (RF grade 2).
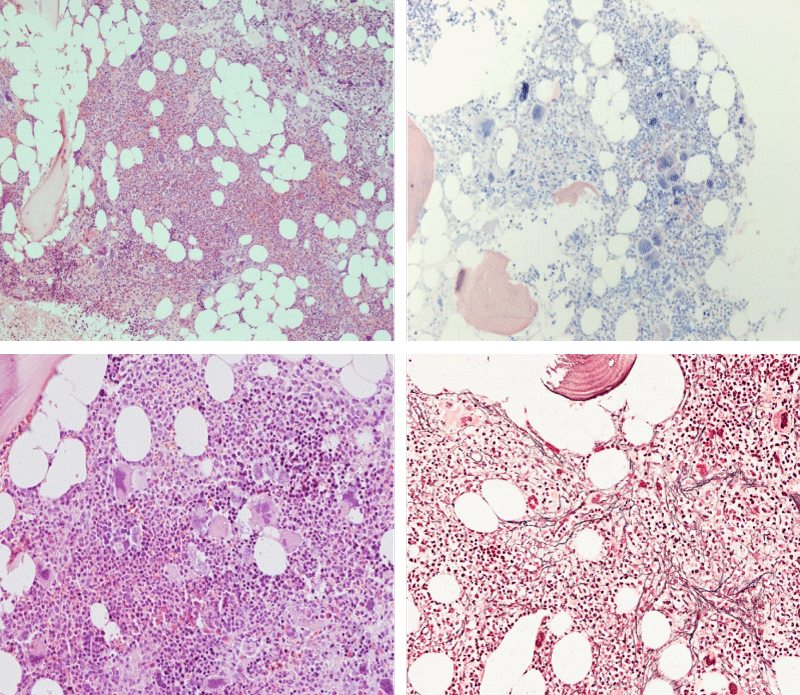
Figure 15: Bone marrow histology of CALR mutated ET/PMGM case 3 after 12 years follow-up at age 51 showing dense clusters of large megakaryocytes with sometimes bulky, cloud-like nuclei in a hypercellular MGM bone marrow with relative reduction of erythropoiesis and increase in reticulin fibers (RF grade 2).
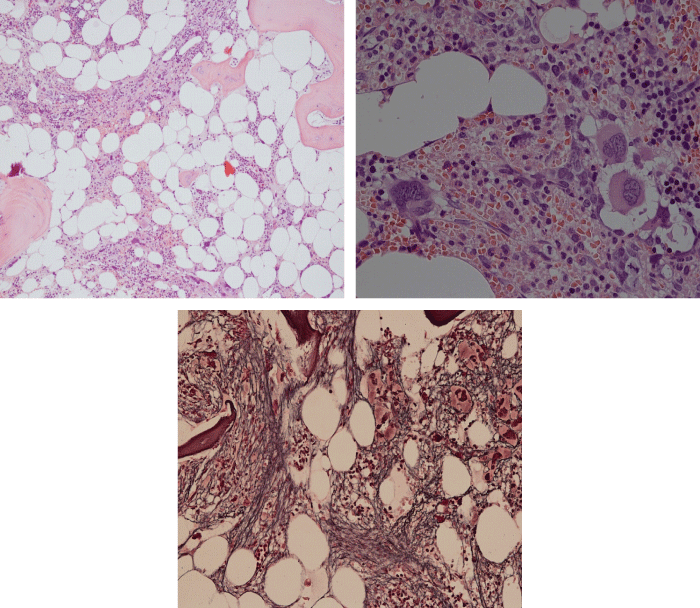
Figure 16: Bone marrow histology of CALR mutated MF/PMGM case 5 (man, 68 years), who first presented with asymptomatic splenomegaly, no constitutional symptoms and symptomatic anemia due CALR advanced myelofibrosis (RF grade 4) in a hypocellular bone marrow showing dysmorphic megakaryocytes in fibrotic parts of the bone marrow. The transfusion dependent anemia responded to EPO, which improved the anemia from a Hb 4.2 →Hb 5,7 mmol/L without the need of blood transfusion during follow-up.
Newly diagnosed JAK2V617F positive ET and prodromal PV patients usually have low serum EPO, increased LAP score, and slight to moderate increased bone marrow cellularity due to an increased megakaryopoiesis (M) in a normocellular bone marrow or present with increased bone marrow cellularity due to increased erythropoiesis and megakaryopoiesis (EM). Bone marrow hypercellularity due to trilinear increase in erythropoiesis, megakaryopoiesis and granulopoiesis (EMG) is the hallmark of classical PV and in advanced JAK2V617F mutated ET (masked PV). This is combined with increased serum LDH levels, high scores for leukocyte alkaline phosphatase stain (LAP score), splenomegaly and high JAK2V617F mutation allele burden between 50% and 100%. Clustered large and giant mature megakaryocyte with hyperlobulated ‘staghorn’ nuclei – which are rare in JAK2V617F mutated MPN - typically present in MPL515 mutated ET patients with no features of PV [30]. The prevalence of MPL515 mutated ET or MF patients ranges from 5 to 10% of the JAK2 wild type MPN population [30].
The Hannover Bone Marrow classification distinguished three MPD disease entities of ET, PV and hypercellular thrombocythemia related to PMGM without features of PV (Table 1) [9,10]. The discovery by Kralovics et al. in 2013 [29], of CALR as the driver cause of JAK2/MPL515 wild type thrombocythemia and PMF led to the second ground breaking event in the molecular landscape of the MPNs that induced in the minds of and extended experiences by Michiels & De Raeve a complete revision and integration of the PVSG/WHO classifications into the current Clinical Laboratory, Molecular and Pathobiological (2018 CLMP) [22-26], criteria for JAK2V617F trilinear MPN and JAK2exon12 PV on the one hand and two distinctive MPL515 and CALR thrombocythemias and myelofibrosis without features of PV on the other hand (Figure 3) [30-32]. CALR mutated thrombocythemia is the third distinct MPN entity featured by bone marrow findings of dysmorphic large megakaryocytes associated with prefibrotic and fibrotic stages of PMGM during lifelong follow-up. This CALR MPN entity has - in contrast to the JAK2-mutated MPNs - no features of PV at time of diagnosis and during follow-up. The clinical and bone marrow histology features of CALR mutated thrombocythemia patients are phenotypically identical to hypercellular ET associated with PMGM defined by Georgii in the Hannover Bone Marrow (BM) Classification (1990) [9] and do belong to the original description of megakaryocytic leukemia (ML) in 1951 by Dameshek [2].
The present study confirms previous reports [9-11,15-18] and recent observations by the Belgian-Dutch-Korean MPN investigators [30-34], that each of the clonal JAK2V617F, CALR and MPL515 mutated ET starts with normocellular ET associated with monilinear megakaryocytic (M) BM proliferation. The natural history of JAK2V617F mutated trilinear MPN runs through subsequent stages of prodromal PV showing dual erythrocytic megakaryocytic (EM) BM proliferation followed by erythrocytic granulocytic megakaryocytic (EMG) BM proliferation in classical, masked and advanced PV during lifelong follow-up. Similarly, CALR driver mutation induces sequential stages of monolinear megakaryocytic (M) BM proliferation in a normocellular bone maarow followed by dual proliferation of megakaryocytic granulocytic (GM) BM proliferation without features of PV. Each of the JAK2V617F, CALR and MPL515 mutated MPNs run through prefibrotic, early fibrotic and advanced fibrotic BM stages during lifelong follow-up.
Authors Contribution
JJM. YK, MK and HDR designed the study. JJM and HDR wrote the manuscript. All authors significantly contributed to clinical and bone marrow data collection.
On behave of the International Collaborations and Academic Research Project on Myeloproliferative Neoplasms: ICAR.MPN, HARMONY MPN Working Party, European Hematology Association (EHA).
- Dameshek W. Physiopathology and coarse of polycythemia vera as related to therapy. JAMA. 1950; 142: 790-797. Ref.: http://bit.ly/2MYKF4n
- Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951; 6: 372-375. Ref.: http://bit.ly/2KDmirl
- Wasserman LR. The management of polycythaemia vera. Br J Haematol.1971; 21: 371-376 Ref.: http://bit.ly/2L8bHUH
- Laszlo J. Myeloproliferative disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD and hemorrhagic thrombocythemia. Semin Hematol. 1975; 12: 409-432. Ref.: http://bit.ly/2KxFiHo
- Berlin NI. Diagnosis and classification of the polycythemias. Sem Hematol. 1975; 12: 339-351. Ref.: http://bit.ly/2RtZrPx
- 2001 WHO classification of the chronic myeloproliferative diseases (CMPD) polycythemia vera, chronic idiopathic myelofibrosis essential thrombocythemia and cMPD unclassifiable. In: Jaffe SS, Harris NL, Stern A, Vardiman JW eds. WHO classification of Tumours of haematopoiesis and lymphoid tissues. Lyon, France IARC. 2001; 31-42.
- 2008 WHO criteria for polycthemia vera, primary myelofibrosis and essential thrombocythemia. Thiele et al In: Swerdlow SH, Campo E, Harris NL et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon France IARC Press. 2008; 40-50.
- Michiels JJ1, Prins ME, Hagermeijer A, Brederoo P, van der Meulen J, et al. Philadelphia chromosome positive essential thrombocythemia and megakaryoblast leukemia. Am J Clin Pathol. 1987; 88: 645-752. Ref.: http://bit.ly/2J4b0sz
- Georgii A, Vykoupil KF, Buhr T, Choritz H, Döhler U, et al. Chronic myeloproliferative disorders in bone marrow biopsies. Path Res Pract. 1990; 186: 3-27. Ref.: http://bit.ly/2L8d7hZ
- Georgii A, Buhr T, Buesche G, Kreft A, Choritz H. Classification and staging of Ph-negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leukemia Lymphoma. 1996; 22: Suppl 1: 15-29. Ref.: http://bit.ly/2WWFB0q
- Michiels JJ. Diagnostic criteria of the myeloproliferative disorders (MPD): essential thrombocythemia (ET), polycythemia vera (PV) and chronic megakaryocytic granulocytic metaplasia (CMGM). Neth J Med. 1997; 51: 57-64. Ref.: http://bit.ly/2Y88Y12
- Michiels JJ, Juvonen E. Proposal for revised diagnostic criteria of essential thrombocythemia and polycythemia vera by the Thrombocythemia Vera Study Group. Sem Thromb Hemostas. 1997; 23: 339-347. Ref.: http://bit.ly/2J0Du6C
- Michiels JJ, Kutti J, Stark P, Bazzan M, Gugliotta L, et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thromboythemia, polycythemia vera and essential megakaryocytic granulocytic myeloproliferation and myelofibrosis. Neth J Med. 1999; 54: 46-62. Ref.: http://bit.ly/2ZCIzbX
- Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol. 2002; 76: 133-145. Ref.: http://bit.ly/2XAjmln
- Michiels JJ, Kvasnicka HM, Thiele J. Doctor’s Brochure 2004, Myeloproliferative Disorders Essential Thrombocythemia, Polycythemia Vera and Chronic idiopathic Myelofibrosis. MPD: Ref.: http://bit.ly/2Ru0TRZ
- Michiels JJ, Hendrik De Raeve, Berneman Z, Van Bockstaele D, Hebeda K, et al. The 2001 World Health Organization and updated European Clinical and Pathological criteria for the diagnosis classification and staging of the Philadelphia-negative chronic myeloproliferative disorders. Sem Thromb Hemostas. 2006; 32: 307-340. Ref.: http://bit.ly/2IyarIC
- Michiels JJ, Berneman Z, Van Bockstaele D, Van Der Planken M, De Raeve H, et al. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Sem Thromb Hemostas. 2006; 32: 174-207. Ref.: http://bit.ly/2YaX6eU
- Michiels JJ, Piche A, De Raeve H, Campr V, Schwarz J. WHO clinical molecular and pathological (WHO-CMP) features of congenital MPLS505N and the acquired MPLW515L/K mutated essential thrombocythemia and myelofibrosis. J Hematol Thromb Dis. 2014; 2: 6. Ref.: http://bit.ly/2L7N3na
- Michiels JJ. Myeloproliferative and thrombotic burden and treatment outcome in thrombocthemia and polycythemia patients. World J Crit Care Med. 2015; 4: 230-239. Ref.: http://bit.ly/2KyB4iX
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H. Changing concepts of diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms: From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol. 2015; 133: 36-51. Ref.: http://bit.ly/2Y4mVgz
- Michiels JJ, Valster F, Wielenga J, Schelfout K, De Raeve H. European vs 2015 World Health Organization clinical molecular and pathological classification of myeloproliferative neoplasms. World J Hematol. 2015; 4: 16-53. Ref.: http://bit.ly/2ZC2szZ
- Michiels JJ, Medinger M, De Raeve H, Schroyens W, Schelfout K, et al. Increased erythrocyte count on top of bone marrow histology, but not by EPO level or JAK2V617F mutation load discriminates between JAK2V617F mutated essential thrombocythemia and polycythemia vera. J Hematol Thromb Dis. 2015; SI.001. Ref.: http://bit.ly/2IY5zM3
- Michiels JJ, Tevet M, Trifa A, Niculescu-Mizil E, Lupa A, et al. 2016 WHO Clinical Molecular and Pathological Criteria for Classification and Staging of Myeloproliferative Neoplasms (MPN) Caused by MPN Driver Mutations in the JAK2, MPL and CALR Genes in the Context of New 2016 WHO Classification: Prognostic and Therapeutic Implications. MAEDICA. 2016; 11: 5-25. Ref.: http://bit.ly/2IY688D
- Michiels JJ, De Raeve H, Valster F, Potters V, Kim Y, et al. Extension of 2016 World Health Organization (WHO) classification and a new set of clinical, laboratory, molecular and pathological criteria for the diagnosis of myeloproliferative neoplasms: from Dameshek to Vainchenker, Green and Kralovics. EMJ. 2017; 2: 72-81. Ref.: http://bit.ly/31NrRbV
- De Raeve H, Fostier K, Valster F, Potters V, Kim Y, et al. Bone Marrow Histology is a Pathognomonic Clue to Each of the JAK2V617F, MPL515 and Calreticulin Mutated Thrombocythemia in Myeloproliferative Neoplasms. Clin Res Hematol. 2018: 1: 1-7. Ref.: http://bit.ly/2IwU3Ih
- De Raeve H, Michiels JJ, Valster F, Potters V, Kim Y, et al. Novel Clinical, Laboratory, Molecular and Pathological (2018 CLMP) Criteria for the Differential Diagnosis of three Distinct JAK2, CALR and MPL Mutated Myeloproliferative Neoplasms: The Role of Driver Mutation Analysis and Bone Marrow Histology. Int J Cancer Res Ther. 2018; 3: 1-12. Ref.: http://bit.ly/2IvwsHY
- James C, Delhommeau F, Marzac C, Teyssandier I, Le Couédic JP, et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia. 2006; 20: 350-353. Ref.: http://bit.ly/2WS1QEA
- Vainchenker W, Constantinescu SN. A unique activating mutation in JAK2 V617F is at the origin of polycythemia vera and allows a new classification of myeloproliferative diseases. Hematology (Am Soc Hematol Educ Progr). 2005; 195-200. Ref.: http://bit.ly/2IZ9JmF
- Klampf T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, et al. Somatic mutations of calreticulin myeloproliferative neoplasms. N Engl J Med. 2013; 369: 2379-2387. Ref.: http://bit.ly/2Rr7iNI
- Kim Y, Park J, Jo I, Lee GD, Kim, et al. Genetic-pathologic characterization of myeloproiferative neoplasms. Exp Mol Med. 2016; 48: 247. Ref.: http://bit.ly/2Ix46go
- Michiels JJ, De Raeve H, Schwarz J, Campr V, Kim Y, et al.. Bone Marrow Histology Characteristics in MPL515 Mutated Thrombocythemia with Various Degrees of Myelofibrosis: A Cross Sectional Follow-up Study in Eight Cases. J Hematol Thrombo Dis. 2018; 6: 2. Ref.: http://bit.ly/2Xn3fHC
- Michiels JJ, Berneman Z, Gadisseur A, De Raeve HD, Schroyens W, et al. Myelofibrosis is a Secondary Event in JAK2 Trilinear Myeloproliferative Neoplasm (MPN) and in CALR and MPL Thrombocythemia: Implications for Novel Treatment Options of Prefibrotic MPN. J Hematol Thromembolic Dis. 2017; 5: 5. Ref.: http://bit.ly/2N4aMHa
- Michiels JJ, De Raeve H. The PVSG/WHO versus the Rotterdam European clinical, molecular and pathological diagnostic criteria for the classifi cation of myeloproliferative disorders and myeloproliferative neoplasms (MPD/MPN): From Dameshek toGeorgii, Vainchenker and Michiels 1950-2018. Int J Bone Marrow Res. 2019; 2: 027-050. Ref.: http://bit.ly/2IvOIBc
- Michiels JJ, Berneman Z, Schroyens W, J ten Kate FW, Lam K, et al. European Clinical Laboratory, Molecular and Pathological (ECMP) criteria for prefi brotic JAK2V617F-Thrombocythemia and Polycythemia Vera versus MPL51- and CALR-Thrombocythemia and Myelofi brosis: From Dameshek to Michiels 1950-2018. Int J Bone Marrow Res. 2019; 2: 001-017. Ref.: http://bit.ly/31SeYxp