
More Information
Submitted: 09 March 2020 | Approved: 30 March 2020 | Published: 03 April 2020
How to cite this article: Michiels JJ, Lam KH, Kate FT, Kim DW, Kim M, et al. Novel European Asiatic Clinical, Laboratory, Molecular and Pathobiological (2015-2020 CLMP) criteria for JAK2V617F trilinear polycythemia vera (PV), JAK2exon12 PV and JAK2V617F, CALR and MPL515 thrombocythemias: From Dameshek to Constantinescu-Vainchenker, Kralovics and Michiels. Int J Bone Marrow Res. 2020; 3: 001-020.
DOI: 10.29328/journal.ijbmr.1001011
Copyright License: © 2020 Michiels JJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Myeloproliferative neoplasms; Essential thrombocythemia; Polycythemia vera; Primary megakaryocytic granulocytic myeloproliferation; Myelofibrosis; JAK2V617F mutation; MPL515 mutation; Calreticulin mutation; JAK2 wild type; Bone marrow histology

Novel European Asiatic Clinical, Laboratory, Molecular and Pathobiological (2015-2020 CLMP) criteria for JAK2V617F trilinear polycythemia vera (PV), JAK2exon12 PV and JAK2V617F, CALR and MPL515 thrombocythemias: From Dameshek to Constantinescu-Vainchenker, Kralovics and Michiels
Jan Jacques Michiels1*, King H Lam2, Fibo Ten Kate2, Dong-Wook Kim3, Myungshin Kim4, Vasily Shuvaev5, Francisca Valster6, Vincent Potters7, Wilfried Schroyens8, Mihaela Andreescu9, Adrian Trifa10, Achille Pich11 and Hendrik De Raeve12
1Hematology and Bloodcoagulation Research Center, Goodheart Institute and Foundation in Nature Medicine & Health, Rotterdam, The Netherlands
2Department of Hematopathology, Erasmus University Medical Center, Rotterdam, The Netherlands
3Leukemia Research Institute, Seoul St Mary’s Hematology Hospital, College of Medicine, The Catholic University of Korea Seoul, South Korea
4Department of Laboratory Medicine, College of Medicine, The Catholic University of Korea and Catholic Genetic Laboratory Center, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul South Korea
5Hematology Clinic in Russian Research Institute of Hematology and Transfusiology, Saint-Petersburg, Russia
6Department of Hematology, Bravis Hospital, Bergen op Zoom, The Netherlands
7Department of Pathology, Bravis Hospital, Bergen op Zoom, The Netherlands
8Department of Hematology, University Hospital Antwerp, Edegem, Belgium
9Romanian Working Group on Myeloproliferative Neoplasm (RWG.MPN) and Department of Hematology, Colentina Clinical Hospital, Bucharest, Romania
10Department of Medical Genetics, “LuliuHatieganu”, University of Medicine and Pharmacy, and Department of Genetics, “Ion Chiricuta”Cancer Institute, Cluj-Napoca, Romania
11Department of Molecular Biotechnology and Health Sciences, Section of Pathology, University of Turin
12Departments of Pathology, OLV Hospital Aalst and University Hospital Brussels, Belgium. On behave of the International Collaborations and Academic Research Project on Myeloproliferative Neoplasms: ICAR.MPN, HARMONY MPN Working Party, European Hematology Association (EHA)
*Address for Correspondence: Dr. Jan Jacques Michiels, Professor, MD, PhD, Goodheart Institute & Foundation in Nature Medicine, Freedom of Science & Education, European Free University Network, Erasmus Tower, Veenmos 13, 3069 AT Rotterdam, The Netherlands, Tel: +86 15757195424; Email: [email protected]
The Myeloproliferative Neoplasms (MPN) of trilinear polycythemia vera (PV) and megakaryocytic leukemia (ML = primary megakaryocytic granulocytic myeloproliferation: PMGM) and Essential Thrombocythemia (ET) in the studies of Dameshek and Michiels are caused by the MPN driver mutations JAK2V617F, JAK2exon12, CALR and MPL515 discovered by Constantinescu-Vainchenker, Green and Kralovics. The JAK2V617F mutated trilinear myeloproliferative neoplasms (MPN) include a broad spectrum of clinical laboratory and bone marrow features in essential thrombocythemia (ET), prodromal PV and erythrocythemic PV, classical PV and advanced stages of masked PV and PV complicated by splenomegaly and secondary myelofibrosis (MF). Heterozygous JAK2V617F mutated ET is associated with low JAK2 allele and MPN disease burden and normal life expectance. In combined heterozygous and homozygous or homozygous JAK2V617F mutated trilinear PV, the JAK2 mutation load increases from less than 50% in prodromal PV and classical PV to above 50% up to 100% in hypercellular PV, advanced PV and PV with MF. Bone marrow histology show diagnostic features of eryhrocytic, megakaryocytic and granulocytic (EMG) myeloproliferation in JAK2V617F mutated trilinear MPN, which clearly differs from monolinear megakaryocytic (M) myelproliferation in MPL and CALR thrombocythemia and dual megakaryocytic granulocytic (MG) myeloproliferation in CALR mutated thrombocythemia. The morphology of clustered large pleomorphic megakaryocytes with hyperlobulated nuclei are similar in JAK2V617F thrombocythemia, prodromal PV and classical PV patients. Monolinear megakaryocytic (M) myeloproliferation of large to giant megakaryocytes with hyperlobulated staghorn-like nuclei is the hallmark of MPL515 mutated normocellular thrombocythemia. CALR mutated thrombocythemia usually presents with high platelet count around 1000x109/l and normocellular megakaryocytic (M) proliferation of immature megakaryocytes with cloud-like hyperchromatic nuclei followed by dual megakaryocytic granulocytic (MG) myeloproliferation followed by various degrees of bone marrow fibrosis. Natural history and life expectancy of MPN patients are related to the response to treatment and the degree of anemia, splenomegaly, myelofibrosis and constitutional symptoms. The acquisition of epigenetic mutations at increasing age on top of MPN disease burden independently predict unfavorable outcome in JAK2V617F, MPL515 and CALR mutated myeloproliferative neoplasms (MPNs, which mutually exclude each other).
The combination of plethoric appearance, splenomegaly, erythrocyte count above 6×1012/L, elevated platelet count and the presence of large megakaryocytes and panmyelosis in the bone marrow is diagnostic for trilinear polycythemia vera [1,2]. Venesection aiming at a haematocrit of 0.40 is the first choice life saving treatment option in newly diagnosed PV that prevents major thrombosis and controls hypervolemic symptoms during long-term follow-up in the majority of PV patients [2-7]. PV is a trilinear erythrocythemic, thrombocythemic and granulocythemic (EMG) myeloproliferation caused by either one unknown bone marrow stimulation factor or the lack of one inhibitory factor, [2,8,9]. Megakaryocyte leukemia (ML) is distinct from PV [10] and has been recognized by Georgii, et al. [11,12], as hypercellular thrombocythemia due to dual chronic or primary megakaryocytic granulocytic myeloproliferation (CMGM/PMGM) without features of PV [13-15].
The Hannover Bone Marrow criteria proposed by Georgii, et al. [11], translated the PVSG criteria in the Hannover BM criteria for ET, PV and PMGM and stages of each by grading of myelofibrosis (MF) as a secondary event in advanced stages of MPDs complicated by anemia, splenomegaly and fibrosis in the bone marrow [11-14,16]. Michiels drew attention to the importance of bone marrow histology as a pathognomonic clue to each of the MPDs ET, PV and PMGM [14-17]. The number and size of mature megakaryocytes in bone marrow biopsies are typically increased in ET and PV. Large megakaryocytes with mature cytoplasm and multilobulated nuclei and the tendency to cluster in small groups close to the sinuses represent the hallmark feature of ET (Figure 2). The histologic background of hematopoiesis in ET at platelet counts above 400x109/L is one of normal cellularity in the early stage [14,17] (Table 3). A slight to moderate increased cellularity due to increased erythropoiesis may be seen in ET with increasing platelet counts between 400 to above 1000x109/L against a background of normally maturing granulopoiesis and eryrhropoesis comparable with the early stage of PV [18-20]. Increase in number and size of clustered large megakaryocytes comparable to ET and a moderate to marked increased cellularity due to increased erythropoiesis/megakaryopoiesis (EM) and erythro-megakaryo-granulopoiesis (EMG) are the diagnostic features of untreated PV [14,17] (Figure 3, Table 4). Increase of large megakaryocytes with mature cytoplasms and multilobulated nuclei in a hypercellular bone marrow is even more conspicuously altered in PV than in ET or early prodromal stage PV. The megakaryocytes in PV usually have a pleomorphic appearance with a wide range of megakaryocyte sizes including small, medium sized and large forms (Tables 3,4) as can be demonstrated in immune stained bone marrow biopsies using monoclonal antibodies against platelet glycoprotein. The characteristic increase and clustering of large megakaryocytes and proliferation of erythropoiesis with hyperplasia of dilatated sinuses are the diagnostic hallmark of untreated PV to distinguish it from secondary erythrocytosis [12,15,17,18], from Ph+ chronic granulocytic leukemia and Ph+ ET [13,15] and most importantly from PMGM [12,15,16]. Bone marrow histology in PMGM is dominated by atypical immature megakaryocytes, which are conspciously large due to increase of nuclear as well as cellular size. The nuclei of megakaryocytes in PMGM are bulky with lobuli becoming clumsy. The lightly stained chromatin and irregular roundish nuclear forms give rise to the co-called cloud-like nuclei, which are almost never seen in ET and PV [11,12,14,16,21].
Within the European Working Group on myeloproliferative Disorders (EWG. MPD founded by Dr. Michiels in 1994). Michiels, et al. [14], translated the Hannover Bone Marrow Classification of Georgii, et al. [11] in a new set of the Rotterdam Clinical and Pathological (RCP) criteria for the diagnosis of ET and PV and chronic, essential or primary megakaryocytic granulocytic myeloproliferation PMGM as the third distinct MPD [11,12,14-17,22] (Table 2). The present appraisal of the myeloproliferative neoplasms (MPN) from Dameshek to Michiels review the clinical laboratory molecular and pathological (CLMP) characteristic of the MPNs caused by the driver mutations JAK2V617F, MPL515, JAK2exon12, CALR and MPL515 discovered by Constantinescu-Vainchenker, Pardani, Green and Kralovics respectively (Tables 1,2, Figure 1).
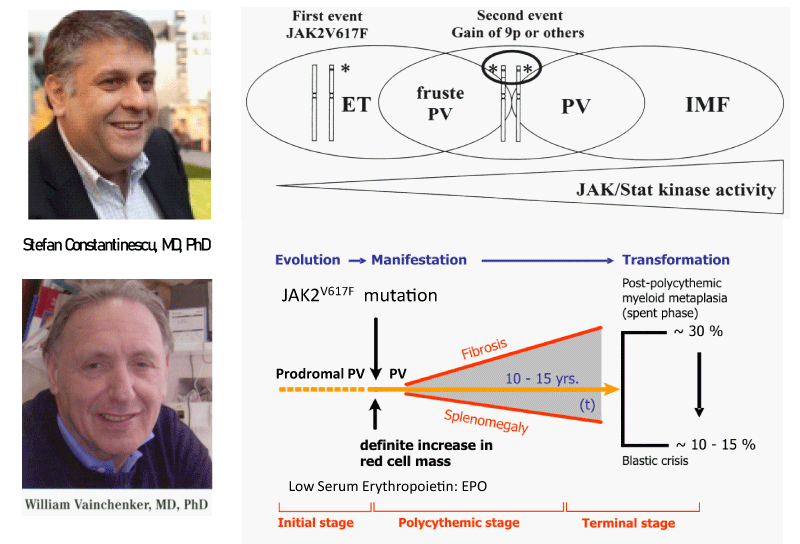
Figure 1: Upper part: The discovery by Constantinescu & Vainchenker of the JAK2V617F somatic mutation as the cause of trilinear myeloproloferative neoplasms and the sequential occurrence of heterozygous JAK2V617F mutation in essential thrombocythemia (ET) and homozygous JAK2V617F mutation in trilinear polycythemia vera (PV) which does explain the occurrence of three sequential phenotypes of ET, PV and myelofibrosis (MF) during life long follow-up in the studies of Dameshek and Michiels. Lower part: Concept of Michiels & De Raeve on the dynamics of the JAK2V617F disease processes in trilinear MPN ranged from normocellular ET and prodromal PV mimicking ET with normal erythrocyte count below 5.8x1012/L to defintive increase in peripheral blood erythrocytes above 5.8x1012/L) in PV followed by masked PV, advanced PV complicated by fibrosis and splenomegaly, spentphase PV and blast transformation of post - PV myelofibrosis. Designedby Michiels 2020.
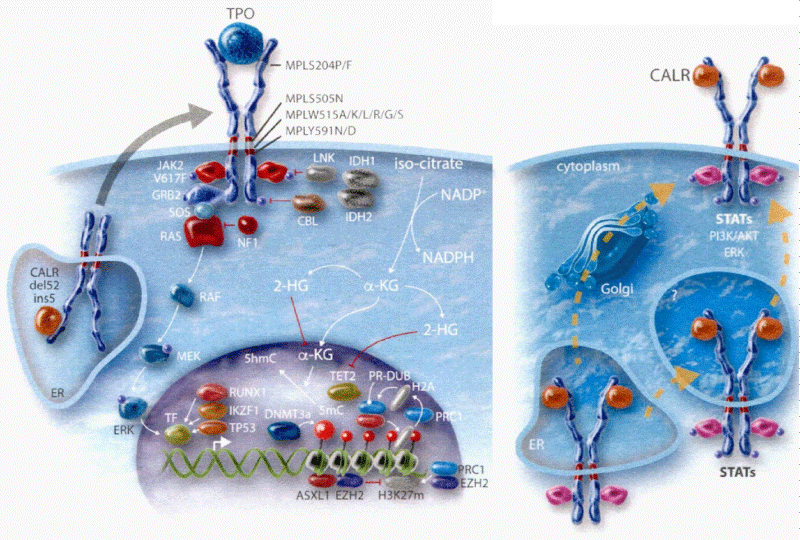
Figure 2: The heterozygous JAK2V617F mutated acquired thrombocythemia and germline TPO and JAK2V671I or JAK2Q534R mutated hereditary thrombocythemia as well as acquired MPL515 and congenital hereditary MPL505 mutated thrombocythemia (ET) are driven by indirect cytokine activation heterozygous JAK2V617F→STAT5, germline JAK2 or TPO→TpoR = MPL. Direct binding of TPO to the D3D4 domain of TpoRor direct cytokine activation (MPL515 and MPL505) cytokine receptor activation induce an ET phenotype of MPN without features of PV (EEC negative) Michiels, et al. 2014 [27]. CALR mutants Type 1 and 2, but not wild type CALR, did induce STAT5 activation via TpoR (MPL) and GCSFR, but not via EpoR. The STAT5 activation via GSCFR was much weak erthan via TpoR (MPL). The extracelllar domain of TpoR (MPL), but not of EpoR, was indispensible for CALR mutant induced activity and the D1D2 distal part of the extracelluar TpoR domain and its associated N-glycosylation sites but not the TPO binding site in the D3D4 domain Araki, et al. Vainchenker and Kralovics 2017) [65].
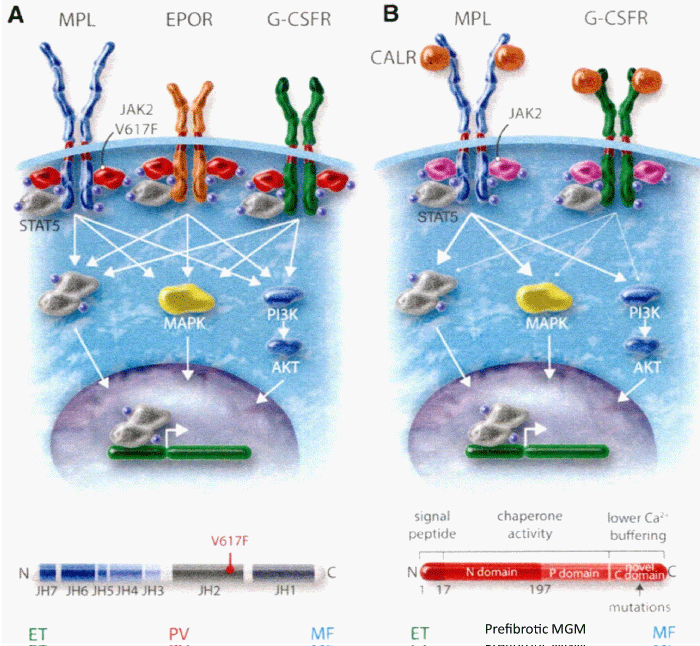
Figure 3: According to Constantinescu-Vainchenker low V617F constitutional kinase activity in heterozygous mutated JAK2V617F mutated patients is enough to produce the ET phenotype via the MPL signalling pathway and that higher V617F constitutional kinase activity in JAK2V617F mutated, heterozygous/homozygous or homozygous mutated patients is needed to produce the sequential stages of prodromal, classical and advanced (masked) PV phenotypes via activation of both the EPO and pathways of hematopietic progenitors cells (Table 1). The JAK2V617F dosage hypothesis has been confirmed at the bone marrow haematopoietic stem cell level by the demonstration that endogenous erythroid colonies (EEC) from ET patients are mainly heterozygous for the JAK2V617F mutation, whereas all PV patients are either hetero/homozygous or mainly homozygous for the JAK2V617F mutation (Figure 8).
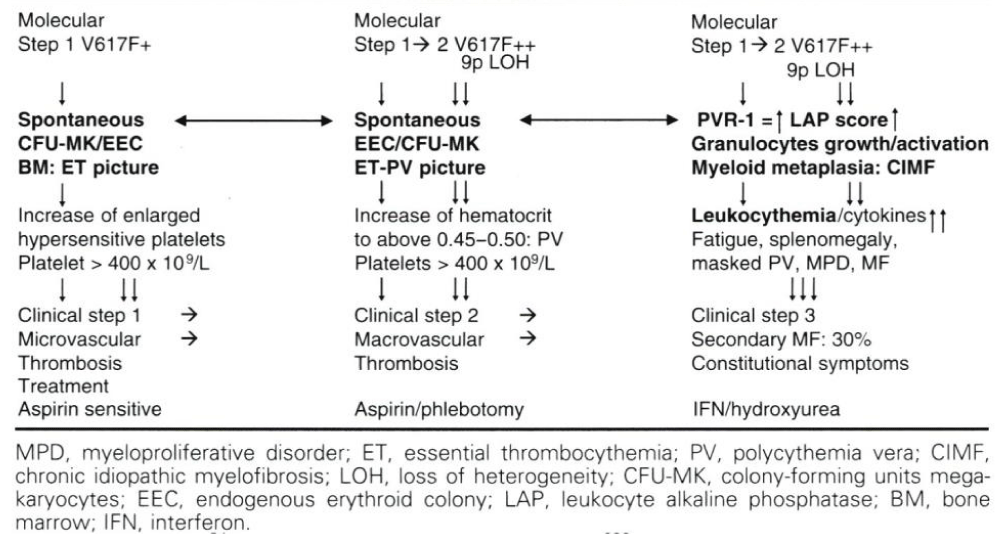
Table 1: The 2006 concept of Michiels (Michiels, et al. 2006ab [8,9], Bellucci & Michiels, 2006 [21] on the molecular etiology of JAK2V617F mutated hypersensitive platelets and platelet-mediated arteriolar erythromelalgic arterial (Michiels, et al. 1984 [86], 1985, 1993) in heterozygous essential thrombocythemia (ET) and major microvascular thrombosis in mixed heterozygous and homozygous or homozygous thrombocythemia and polycythemia vera (TPV) complicated by splenomegaly due to myeloid metaplasia of the spleen and secondary bone marrow fibrosis according to the dosage hypothesis of Constantinescu & Vainchenker (James, et al. 2005 [57], Vainchenker & Constantinescu, 2005 [59], Villeval, et al. 2006 [60]).
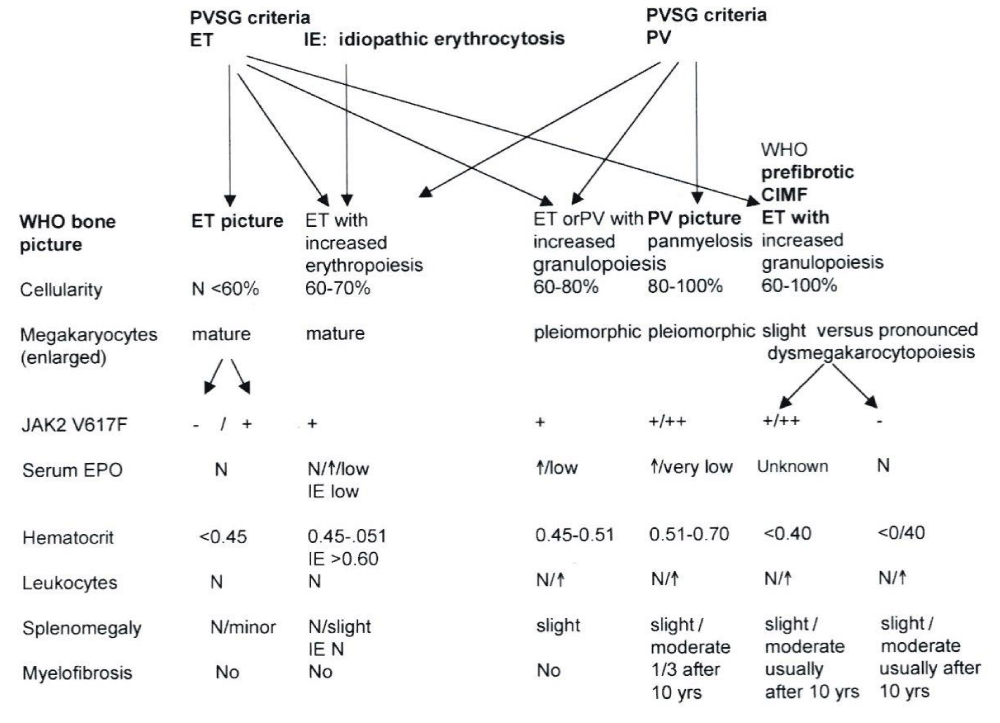
Table 2: The spectrum of JAK2V617F mutated ET, PV and MF versus JAK2 wild type normocellular ET and hypercellular ET associated with prefibrotic PMGM [11,12,15] according to ECP criteria [8] by intergrating the PVSG/WHO bone marrow features into the ECP and ECMP criteria of myelproliferative Disorders [15,17], MPD Doctor’s Brochure 2004, Michiels, et al. [8,9]).
| Table 3: Clinical, Laboratory, Molecular and Pathobiology (2015-2020 CLMP)criteria for diagnosis of JAK2V617F mutated essential thrombocythemia (ET). | |
| Clinical, laboratory, and molecular (CLM) criteria | Bone marrow Pathology (P) criteria |
| Prefibrotic ET | Normocellular ET |
| 1. Platelet count of > 350 x109/l 2. Heterozygous JAK2-V617F mutation, and low JAK2 allele mutation load 3. Normal erythrocytes < 5.8x1012/L males, < 5.6 x1012/L females 4. Hemoglobin (Hb) and hematocrit (Ht) normal or upper range of normal |
Normocellular bone marrow (< 60%), Megakaryocytic (M) proliferation of clustered of medium sized to large (pleomorphic) mature megakaryocytes in anormocellular bone marrow (< 60%), no proliferation of erythropoiesis and granulopoiesis. Reticuline fibrosis (RF) 0 or 1 |
| Prefibrotic prodromal PV | ET with bone marrow features of PV |
| 1. Platelet count of > 350 x109/l. Hb and Ht in upper range of normal, but erythrocyte count < 5.8x1012/L males, < 5.6x1012/L females. 2. Presence of JAK2-V617F mutation and variable JAK mutation load 3. Low serum EPO, increased LAP score |
Increased cellularity (60%-80%) due to increased erythrocytic, megakaryocytic (EM) proliferation or trilinear erythrocytic, megakaryocytic, granulocytic (EMG) proliferation. Increase of clustered medium sized to large (pleomorphic) mature megakaryocytes. Spontaneous EEC. RF 0 or 1 |
| Prefibrotic hypercellular ET | EMG, masked PV and MG fibrotic stages |
| 1. Platelet count of > 350 x109/l, 2. Presence of JAK2-V617F mutation and high JAK2 mutation load 3. Moderate myeloid neoplasia of the spleen ®splenomegaly 4. No preceding or allied CML, PMGM, RARS-T or MDS . ------------------------------------------------- Clinical stage 1: Hb and Ht in lower range of normal: hb> 12 g/dl, normal LDH and CD34+ Clinical stage 2: anemia Hb < 12 to > 10 g/dL, LDH, and splenomegaly Clinical stage 3: severe anemia, Hb <10 g/dL, LDH, CD34+, leukoerythroblastose, tear drop erythrocytes, and large spleen |
Hypercellular due to increased erythrocytic, megakaryocytic and granulocytic myeloproliferation (EMG, masked PV, prefibrotic) or increased megakaryocytic, granulocytic (MG, fibrotic) proliferation with relative reduced erythroid precursors. Loose to dense clustering of more pleiomorphic megakaryocytes with hyperploid or clumpsy nuclei Grading of reticulin fibrosis (RF, [113]) and myelofibrosis (MF, [11,12,109]) -Prefibrotic: RF- 0/1 = MF-0, no/minor splenomegaly -Early fibrotic EMGM: RF 2 = MF 1 and minor or moderate splenomegaly -Fibrotic EMGM: RF3, RCF = MF2 and overt splenomegaly Post-ET MF: RF3/4 = MF-2/3 (WHO criteria) |
| Table 4: Clinical, Laboratory, Molecular and Pathobiology (2015-2020 CLMP) criteria for the diagnosis of prodromal, masked and classical JAK2 mutated polycythemia vera (PV) versus primary or secondary erythrocytoses | |
| Clinical, laboratory, molecular (CLM) criteria | BonemarrowPathology (P) criteria |
| Major criteria for PV A 1. Erythrocytes > 5.8x1012/L males and > 5.6x1012/L females. Hemoglobin and hematocrit upper range of normal or increased. A 2. Heterozygous and/or homozygous JAK2V617F or JAK2exon12 mutation. A 3. Low serum Epo level. Confirmative criteria B 1. Persistent increase of platelet count x109/L: grade I: 400-1500, grade II: > 1500. B 2. Granulocytes > 10 x109/l or Leukocytes > 12 x109/l and raised LAP-score or increased CD11b expression in the absence of fever or infection. B 3. Myeloid neoplasia of the spleen ® splenomegaly on ultrasound echogram (> 12 cm length in diameter) or on palpation. B 4. Spontaneous endogenous erythroid colony (EEC) formation (optional). |
PV. Increased cellularity (60% - 100%) due to increased erythocytic, megakaryocytic (EM) proliferation or trilinear erythrocytic, megakaryocytic and granulocytic (EMG) proliferation. Increase of clustered medium to large (pleomorph) megakaryocytes with hyperlobulated nuclei. Hypercellular (90% - 100%) due to EMG prefibrotic advanced or masked PV with predominant increased fibrotic megakaryocytic, granulocytic (MG) proliferation with relative reduced erythroid precursors. Erythrocytosis. Normal erythropoiesis, normal granulopoiesis and megakaryocytes of normal size, morphology and no clustering. Grading of secondary reticulin fibrosis (RF, [113]) and myelofibrosis (MF) [11,12,19]. Prefibrotic: RF-0/1 = MF-0; Early fibrotic: RF-2 = MF-1; Fibrotic: RCF 3 = MF-2; Post-PV MF: RF 4 = MF-3. |
Diagnostic differention of ET and PV by erythrocyte count and BM histology
According to Dameshek, [2], Georgii, et al. [11,12], and Michiels, et al. [6,7,14-16,21,23-36], the diagnosis of ET according to PVSG and WHO criteria is one of exclusion. Bone marrow and erythrocyte count were not used in the PVSG/WHO classification as a specific clue to ET in various MPNs [6,7,15-17,31,32]. Wassermann, et al. [37], introduced crude inclusion criteria for the PVSG-01 randomized clinical trial to be sure that patients do have PV because they were subjected to potential leukemogenic agents P32 and chlorambucil as compared phlebotomy aiming at a hematocrit to below 0.50 [37]. These crude criteria are used by the PVSG since 1975 as diagnostic criteria for PV [38]. The PVSG [38] and 2008 and 2016 WHO criteria for PV [39-41] did not measure erythrocyte counts and MCV and did not use bone marrow histology features and persisted to use only crude cut-off levels for hemoglobin and hematocrit (Hb> 18.5 g/dl and Ht> 0.60 in men and Hb> 16.5 and Ht> 0.56 in women) as surrogate measures of red cell mass (RCM) to separate ET from PV.
Michiels, Thiele & De Raeve used bone marrow histology and erythrocyte and platelet counts as pathognomic clue to distinguish all variants of MPN from reactive thrombocytosis, BCR/ABL positive thrombocytheia in chronic myeloid leukemia (CML), and thrombocythemia in myelodysplastic syndromes (MDS, 5q minus syndrome) by demonstrating that clustered mature large megakaryocytes occur in MPN, small monolobulated megakaryocytes in CML and dysmorphic megakaryocytes in MDS [6-8,14,15,23,31,32,34,42].
Megakaryocytes are identical large, mature and pleiomorphic in prefibrotic JAK2V617F positive ET and PV patients (Tables 2-5) and clearly different from the large giant mature megakaryocytes in MPL thrombocythemia (Table 6) and from the large immature megakaryocytes with ‘cloud-like’ nuclei in CALR positive thrombocythemia (Table 7).
| Table 5: 2015-2020 CLMP staging of JAK2V617F positive prodromal PV, erythrocythemic PV, classical PV, masked PV and MF, inapparent PV, spent phase PV and post-PV myelofibrosis (MF). | |||||||
| PV: CLMP stage | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
| Clinical Diagnosis | Prodromal PV | Erythrocythemia (E) | Early PV | Classical PV | Masked PV Inapparent PV |
Advanced PV Spent Phase PV |
Post-PV MF |
| LAP-score, CD11B | | | | | / | | Variable |
| EEC | + | + | + | + | + | + | + |
| Red Cell Mass | N | N | | / | / | ¯ or | Variable |
| Erythrocytes x1012/l | < 5.8 | > 5.8 | > 5.8 | > 5.8 | N | N | ¯ |
| Leukocytes x109/l | < 10 | < 10 | 10 - 12 | 12 -15 | >15 | or | > 20 |
| Platelets x109/l | > 400 | < 400 | < or > 400 | > 400 | +1000 | or | Variable |
| CLMP bone marrow histology |
EM | EM | EM | EMG | EMG | MG-MF | MF |
| BM cellularity (%) Grading RF Grading MF57 |
50-80 RF 0-1 MF 0 |
50-80 RF 0-1 MF 0 |
60-100 RF 0-1 MF 0 |
80-100 RF 0/1, MF 0 |
80-100 RF 1/2 MF 1 |
60-100 RF 3/4 MF 2/3 |
¯ RF 3/4 MF 2/3 |
| Spleen size: | |||||||
| On echogram Below MCL |
< 12-15 0-3 |
<13 NP |
12-15 0-3 |
12-16 4-6 |
18-> 20 > 6 |
16 > 20 > 6 |
> 20 cm > 8 cm |
| JAK2V617F load Granulocytes % |
Low +(++) |
low +(++) |
Moderate < 50% + | Mod/High + / ++ |
High > 50% ++ | High >50% ++ |
High > 50% ++ |
| Risk stratification ®Therapeutic implications |
Low Aspirin |
Low Phlebot Aspirin |
Low Phlebot Aspirin |
Intermediate IFN |
High IFN if non Responsive HU-JAK2 inh |
JAK2 inhibitor | Post-PV MF |
| * = increased, ¯ = decreased, N = normal, + = present or heterozygous; ++ = homozygous, RCT = randomized clinical trial. | |||||||
| Table 6: 2015-2020 Clinical Laboratory, Molecular and Pathobiology (CLMP) criteria for the diagnosis of normocelular ET carrying one of the MPL515 mutations. | |
| Clinical, laboratory, molecular (CLM) criteria | Bone marrow Pathology (P) criteria |
| 1. Platelet count > 350x109/L and presence of large platelets in blood smear. 2. Normal Hemoglobin, haematocrit and erythrocyte count. 3. Presence of MPL515 mutation. 4. Normal serum EPO. 5. Normal LAP score (CD11b). 6. No or slight splenomegaly. 7. No preceding or allied CML, PV, PMGM, RAS-T or MDS. Clinical staging similar as in CALR thrombocythemia based on the degree of anemia, splenomegaly and myelofibrosis. |
Megakaryocytic (M) proliferation in a normocellular (< 60%) bone marrow featured by large to giant mature megakaryocyte with hyperlobulated, staghorn-like nuclei. No increase of erythropoiesis, and granulopoiesis. No or slight increase in reticulin RF 0/1. Grading of bone marrow fibrosis: reticulin fibrosis (RF), Wilkins, et al. [113] and myelofibrosis (MF), Georgii, et al. [11,12], Thiele, et al. [109]. |
| Table 7: Clinical Laboratory, Molecular and Pathobiology (2015-2020 CLMP) criteria for hypercellular thrombocythemia associated with primary megakaryocytic, granulocytic myeloproliferation (PMGM) caused by calreticulin (CALR) mutations. | |
| CLM criteria CALR thrombocythemia. | Bone marrow Pathology (P) criteria. |
| A1 No preceding or allied other subtype of myeloproliferative neoplasm PV, CML, MDS. The main presenting features is pronounced isolated thrombocythemia with platelet count around or above 1000x109/L. A2 CALR mutation and JAK2 wild type. C Clinical stages of CALR Thrombocythemia. C 1. Early clinical stage: Hb > 12g/dL, slight to moderate splenomegaly, thrombocytosis around or above 1000x109/L, normal LAP score. C2. Intermediate clinical stage: slight anemia Hb < 12 to > 10 g/dL, decreasing platelet count, splenomegaly, increased LDH and definitive tear drop erythrocytes. C3. Advanced stage: anemia Hb < 10 g/dL, tear drop erythrocytes, increased LDH, increased CD34+ cells, pronounced splenomegaly, normal or decreased platelet counts, leucocytosis or leukopenia. |
Megakaryocytic (M) myeloproliferationof dense clustered atypical large immature megakaryocytes with hypolobulated nuclei in a normocellular bone marrow. Prefibrotic dual megakaryocytic granulocytic (MG) myeloproliferation and relative or absolute reduction of erythropoiesis and erythroid precursors. Abnormal dense clustering and increase in atypical medium sized, large to giant immature megakaryocytes containing bulbous (cloud-like) hypolobulated nuclei and definitive maturation defects No features of PV in blood and bone marrow. Grading reticulin fibrosis (RF), Wilkins, et al. [113], and myelofibrosis (MF) Georgii, et al. [11,12], Thiele, et al. [109]. MF 0 Prefibrotic CALR MG, no reticulin fibrosis RF 0/1. MF 1 Early fibrotic CALR MG slight reticulin fibrosis RF 2. MF 2 Fibrotic CALR MG increase RF grade 3 and slight to moderate collagen fibrosis. MF 3 Advanced fibrotic CALR MG with collagen fibrosis-osteosclerosis. |
Erythrocyte count above the upper limit of normal (> 5.8 x10/12L in males and > 5.6 x10/12L in females) on top of characteristic bone marrow histology obviates the need to measure RCM [5,16,27,31,32,43,44] (Figure 2, Table 3). Bone marrow histology of sequential stages in prodromal, overt and advanced PV is typically featured by increased cellularity due to increase erythrocytic megakaryocytic (EM), erythrocytic, megakaryocytic granulocytic (EMG), and predominant megakaryocytic granulcytic (MG) myeloproliferation (Figures 5,6 and Tables 4,5). PV is frequently preceded by ET or prodromal PV for several up to more than 10 years. Bone marrow iron stain is usually positive in normocellular ET, but negative in PV. At red cell mass (RCM) values above 30 ml/kg the erythrocytes are above 5.8x1012/L which is diagnostic for PV in all 19 CLMP defined PV patients [33] (Figure 4). The CLMP defined ET had normal RCM and erythrocyte counts below 5.8 x1012/L with hematocrit values ranging from 0.40 to 0.45 (Figure 1, Tables 2-4). At erythrocytes above 5.8 x1012/L, diagnostic for PV, (Table 4), the hematocrit values ranged from 0.46 to 0.72 and Hb values ranged from 15.0 to 20.9 g/dL, which are clearly below the 2008/2016 WHO-defined criteria for PV [39,41,45]. Seven ET patients had normal RCM at erythrocyte counts between 4.4 to 5.3 x1012/L of whom 4 had normocellular (< 60%) ET and 3 had hypercellular (60%-80%) prodromal PV bone marrow histology. Erythrocyte counts remain above 5.8x1012/L in phlebotomy induced PV in hematological remission due to microcytic erthrocytosis (MCV below 70 fl) caused by iron deficiency [2,10] (Figure 4). In newly diagnosed PV patients the state of PV in remission by phlebotomy due to iron deficiency induced microcytosis of erythrocytes is reached after about 1 to 2 years of repeated venesections (Figures 3,4). This is associated with the relief of hypervolemic symptoms in PV patients during long-term or even life-long follow-up [2,4,31,32].
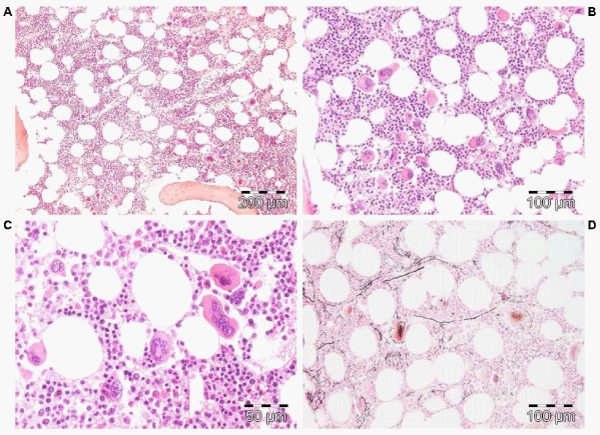
Figure 4: Megakaryocytic (M) proliferation in a normocellular bone marrow (60%) with increase of large mature megakaryocytes, multilobulated nuclei and the tendency to cluster in small groups close to the sinuses is the hallmark of JAK2V617F mutated ET. Pathology Laboratory Lam Rotterdam and De Raeve, Brussels.
In the late 1970s, the London PV study Group of Pearson, Messinezy, Thomas and Weitherley-Mein demonstrated that on top of the microvascular disease of thrombocythemia, the incidence of major arterial and venous episodes in PV correlated positively with increased haematocrit level [46-49]. A mean haematocrit of 0.60 and a mean platelet count of 512x109/L at time of diagnosis of PV were associated microvascular ischemic events and major thrombosis in 49%. The risk of major vascular episodes were the lowest at hematocrits below 0.44, higher at hematocrits above 0.45 and the highest at hemotocrits above 0.50 as was the case in the PVSG 01 study when not on low dose aspirin for the prevention of platelet-mediated microvascular ischemic distubances, TIAs and acute coronary syndromes [36,50,51]. Low dose aspirin at hematocrits of around 0.40 in JAK2V617F mutated ET and PV significantly reduces the incidences of both microvascular as well as major vascular events as compared to not using aspirin in randomized clinical trials [30,36,52]. Phlebotomy on top of low dose aspirin (40 to 80 mg OD) is the cornerstone of treatment of newly diagnosed PV patients with low, intermediate and high MPN disease burden [34-36,53].
JAK2V617F mutated trilinear MPN
EPO-independent progenitor colony-forming unit-erythroid (CFU-E) and burst forming unit-erythroid (BFU-E) labelled as spontaneous endogenous erythroid colony formation (EEC) became the hallmark of PV [54]. Analysis of about 500 PV patients from 26 studies indicated that EEC in expert hematological laboratories has a near 100% diagnostic specificity for overt and masked PV and ET mimicking PV or latent PV [54,55].
The 9p loss of heterogeneity (9pLOH) due to mitotic recombination of chromosome 9p is the most frequent chromosomal lesion described in PV (∼33%) not detectable by cytogenetic analysis [56]. Kralovics, et al. sequenced 19 candidate genes mutations within the 9pLOH region and no mutations were found in the Janus kinase (JAK) gene, but Kralovics did not screen the JH2 pseudokinase gene thereby overlooking the JAK2V617F in the JH2 pseudokinase gene (Figure 1). Constantinescu & Vainchenker searched for a mutation in the 9pLOH region of the complete JAK2 gene and did found the JAK2V617F in the JAK2 pseudogene of 3 PV and 2 controls by detection of a G-to-T mutation at nucleotide 1849 in exon 12 leading a substitution of valine to phenylalanine at position 617 (V617F) in the JAK2 pseudo-gene. This V617F substitution of the JAK2V617F mutation was present in 40 of 45 PV patients, in 9 of 21 ET patients and in 3 of 7 MF patients [57], (Figure 1). The JAK2V617F substitution was absent in patients with secondary erythrocytosis (N = 35) and 15 controls [57]. Thirty percent of PV patients are homozygous for JAK2V617F mutation without socalled 9pLOH due to mitotic recombination, whereas heterozygous JAK2V617F mutated ET and PV patients showed the presence of 9pLOH [57,58] (Table 1, Figure 1).
Constantinescu & Vainchenker, [59] demonstrated that the acquisitions of heterozygous, hetero-homozygous and homozygous due to mitotic recombination JAK2V617F mutation on chromosome 9p are the driver causes of sequential megakaryocytic (M), erythrocytic megakaryocytic (EM) and erythro-megakaryo-granulocytic (EMG) myeloproliferations seen in normocellular ET, prodromal PV and classical and advanced PV in trilinear MPN (Figures 5,6), [21,57,60], (Table 1, Figure 1). The JAK2V617F mutation as the driver cause of trilinear MPN was immediately confirmed in three large groups of ET, PV and MF patients due to inside information during the peer review process in 2004 [58,59,61]. JAK2V617F mutation induces a loss of inhibitory activity of the JAK2 pseudokinase part on the JAK2 JH kinase part leading to enhanced activated of the normal JAK2 JH1 kinase activity, which makes the TPO, EPO and granulocyte growth factor receptors on the hematopoietic progenitor cells hypersensitive to their growth factors TPO, EPO and granulocyte growth factor (Figures 5,6). The JAK2V617 mutated hematopietic cells produce Contitutively activated platelets and leukocytes (increased leukocyte alkaline phosphatase: LAP) and quantitative increase of platelets erythrocytes and granulocytes (Table 1). The sequential occurrence of low heterozygous, combined heterozygous and homozygous and high homozygous JAK2V617F allele load can readily explain the sequential occurrence of ET, prodromal PV, classical PV and advanced PV followed by secondary MF in trilinear MPN during lifelong follow-up. JAKV617F trilinear MPN is clearly differentfrom JAK2 wild type hypercellular ET associated with PMGM [11,12,14,16] as the third distinct entity of MPD without features of PV (Figure 2, Tables 12-15 in Michiels, et al. [8]). According to 2006 ECMP criteria the sequential transitional states of JAK2V617F disease entity ranged from heterozygous normocellular ET and latent PV mimicking ET labelled as prodromal PV or forme fruste PV followed by heterozygous/homozygous mutated erythrocythemic and early PV, and homozygous nutated advanced PV and post-PV myelofibrosis (Figures 1,2, Table 1), [8,21,62]. The JAK2V617F mutated trilinear MPN phenotypic expression includes normocellular ET, prodromal PV, erythrocythemic PV with normal platelet and leukocyte count, classical PV, masked PV, and various degrees of splenomegaly and myelofibrosis (MF) [57,59] (Figure 1, Table 1). The quantitative JAK2V617F allele burden in neutrophils and in CD34+ cells from the same blood sample in 96 JAK2-positive MPN patients (17 ET, 64 PV and 15 MF mean follow-up 7 years) were below 50% in the majority of ET and about half of the PV patients [63]. The JAK2V617F- CD34+ allele burden were above 50% in about half of the PV patients and the majority of MF indicating advanced trilinear MPN disease burden. The neutrophil JAK2V617F allele burden is usually below 50% in ET and above 50% in PV and MF. The CD34+ allele burden is much lower than the neutrophil JAK2V617F allele burden in ET and early stage PV with no splenomegaly [64]. This is completely in line with the concept that some maturation of JAK2V617F mutated hematopoietic stem cells is needed to neoproliferate because of hypersensitivity megakaryopoesis to TPO and erythropiesis to EPO [64,65] (Figures 5,6, Table 1). The neutrophil JAK2V617F allele burden alone can overestimate the MPN disease burden at the bone marrow progenitor cell level in early stage ET and PV neoproliferative disease [63,64].
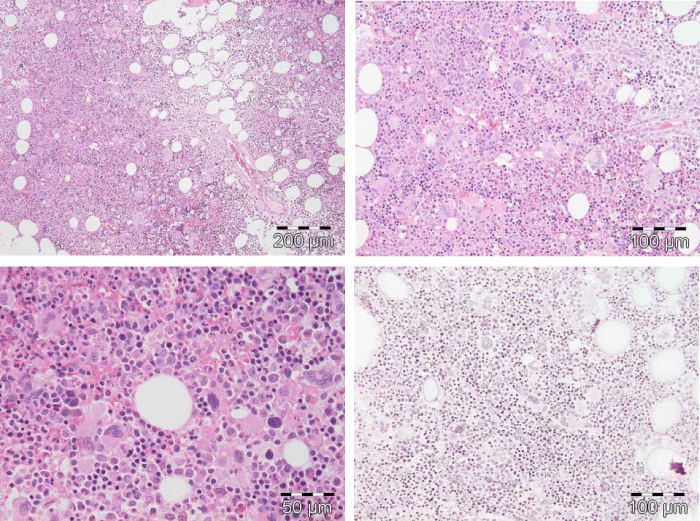
Figure 5: Typical trilinear bone marrow histology [2,18] in classical polycythemia vera (PV) with increased cellularity (100%) due to increased erythocytic, megakaryocytic (EM) proliferation or trilinear erythrocytic, megakaryocytic and granulocytic (EMG) proliferation. Increase of clustered medium to large (pleomorph) megakaryocytes with hyperlobulated nuclei (Pathology Laboratory Rotterdam Lam and Brussels, Dr De Raeve).
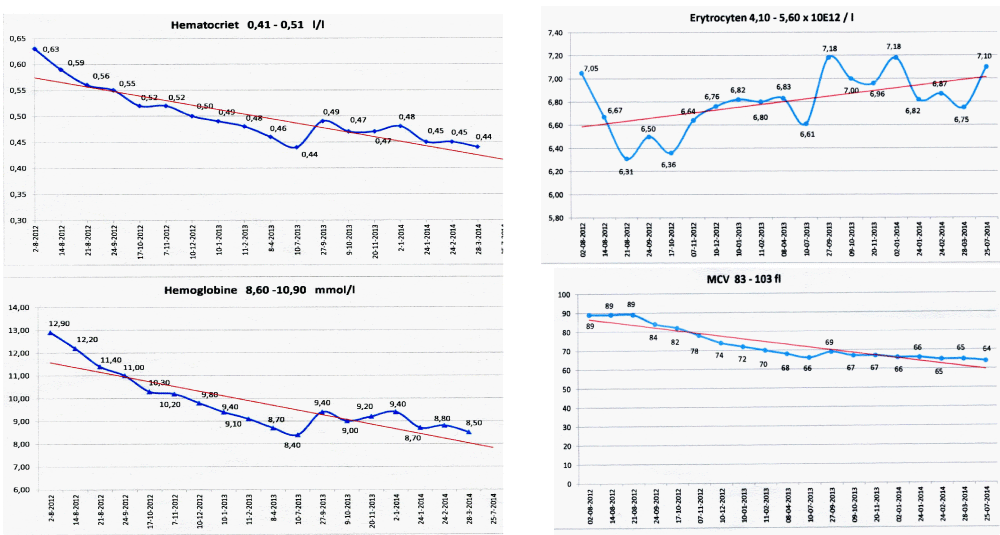
Figure 6: State of the art treatment according to Dameshek [2,4] of a newly diagnosed PV patient (Ht 0.63, Hb 12.9 mmol/L, erythrocytes 7.1 x1012/L and MCV 89 fL) with repeated vene sections as confirmed by Pearson & Wetherley-Mein [48] and Messinazy, et al. [49] of the London PV Study Group [46,47]. Repeated venesections for more than 1 year (from August 2012 to September 2013) was needed to induce a complete hematological remission (CHR) reaching the desired plateau of Ht 0.45, Hb 9.0 mmol/L, and MCV of 66 fL. While on low dose aspirin neither microvascular nor major thrombosis did occur. Once the iron deficiency state is reached the erythrocytes remain microcytic and reach values of 7.0 to 7.2x1012/L without further need of phlebotomy due to the persistence of the iron deficienct state [2,30]. Correction of the Ht from 0.63 to below 0.45 is associated with reduction of major venous and arterial thrombotic events [8,9,48,51], but the microvascular thrombotic syndrome of associated thrombocythemia persisted. Low dose aspirin in ET in the Dutch Collaborative Low dose Aspirin in Thrombocythemia (Dutch CLAT) Van Genderen studies [89-91,99] and low dose aspirin on top of phlebotomy or hydroxurea in the European Collaborative Low dose Aspirin in PV (ECLAP, [50]) reduced the incidences of microvascular disturbances and major thrombosis from above 50% per 100 pt/yr to less than 3% per 100 pt/yr in both ET and PV [91,94], but does not prevent the progression of JAK2 mutated MPN disease in terms of JAK2 mutation load, MPN disease burden like progressive leukocythemia, thrombocythemia, splenomegaly and constitutional symptoms [6,7,34].
Bone marrow morphology of clustered medium to large mature megakaryocytes is similar in heterozygous-mutated JAK2V67F ET and homozygous-mutated JAK2V617FPV patients at time of diagnosis (Figures 2,3, Tables 3-5), [8,9,13,24,25,66]. The bone marrow in heterozygous JAK2V617F ET is normocellular with increased clustered large megakaryocytes (M) proliferation and no or slight increase of erythropoiesis [8,9,24,61,66-68]. The bone marrow in JAK2V617F PV patients with less than 50% mutation load is hypercellular (60-80%) due to increased erythropoiesis and megakaryopiesis in prodromal PV. Classical, masked and advanced stages of PV typically show a trilinear 90-100% hypercellular bone marrow [2] due to increased erythro-megakaryo-granulocytic (EMG, [15,17,18]). The JAK2V617F allele burden in WHO-defined advanced PV and post-PV myelofibrosis(MF) patients ranged from 50% to 100% [63,67,69-71]. According to the Vainchenker’s “dosage” concept, heterozygosity for the JAK2V617F mutation in acquired ET and autosomal dominant JAK2 or TPO mutated hereditary ET (HET) is enough to activate megakaryocytes to induce the ET clinical phenotype [8,21,27,28,36,60,72] (Figure 5, Table 1). Patients with dominant hereditary ET (HET) heterozygous for the JAK2V617I and JAK2R564Q germ line mutations have a clinical ET phenotype with normal values for Hb, Ht, erythrocytes, thrombopoietin (TPO), and erythropoietin (EPO) levels. The response to EPO in the EEC assay was normal in congenital JAK2V617I and JAK2R564Q [73-75], but increased in acquired JAK2V617F mutated ET and PV (Figure 5, Table 1) [8]. According to the JAK2V617F dosage hypothesis the JAK2V617F mutation load is low in heterozygous mutated ET and increases from below to above 50% inpatients with homozygous JAK2V617F mutated PV, advanced PV and post-PV myelofibrosis [8,58,64,76] (Figure 6). According to Vainchenker’s “dosage” concept the higher intracellular levels of JAK2V617F kinase activity in homozygous mutated progenitor stem cells preferentially activate the erythropoietin receptor (EPOR) and generate a PV-like phenotype with erythrocytes above 5.8x1012/L and increased activated platelet and leukocyte counts (Figures 6, Table 1) [8,9,24,35,36,57,60,70,72].
The JAK2V617F allele burden was directly correlated with increased levels of hematocrit, neutrophil count, LDH and leukocyte alkaline phosphatase (LAP) score, spleen size on echogram [8] (Table 1) and with decreased values for platelets, serum ferritin, and erythropoietin, with higher relative risks for aquagenic pruritus, spleen size on echogram, total thrombosis and the need for myelosuppressive treatment [69]. The JAK2V617F allele burden in granulocytes in a prospective study of 175 PV patients could be quantified as 1%-25%, 25% to 50%, 50%-75% and 75%-100% in 57, 50, 34 and 32 PV patients respectively [69]. Prefibrotic heterozygous JAK2V617F mutated ET usually runs a benign course with low JAK2 and MPN burden and a normal to near normal life expectancy (Table 1) [8,9,21] Figure 7, [76]. Prefibrotic hetero/homozygous JAK2V617F mutated prodromal and classical PV usually have a low mutation burden associated with microvascular and major thrombosis at time of presentation (Table 1). Homozygous JAK2V617F mutated trilinear PV result in high JAK2V617F and hypercellular MPN burden associated with progressive extramedullary myeloid neoplasia of the spleen (MNS), splenomegaly and cytokine mediated MF during long-term follow-up [70,76] (Table 1, Figure 6). Transition of heterozygous into homozygous JAK2V617F mutation due to mitotic recombination of chromosome 9p (9pLOH) is strongly correlated with progression into advanced PV and masked PV with splenonomgaly in figures 1 and 2 [77], Table 4, [31,32] followed by post-PV myelofibrosis and associated with high JAK2 allele burden [6,7,24,34,63,70,76].
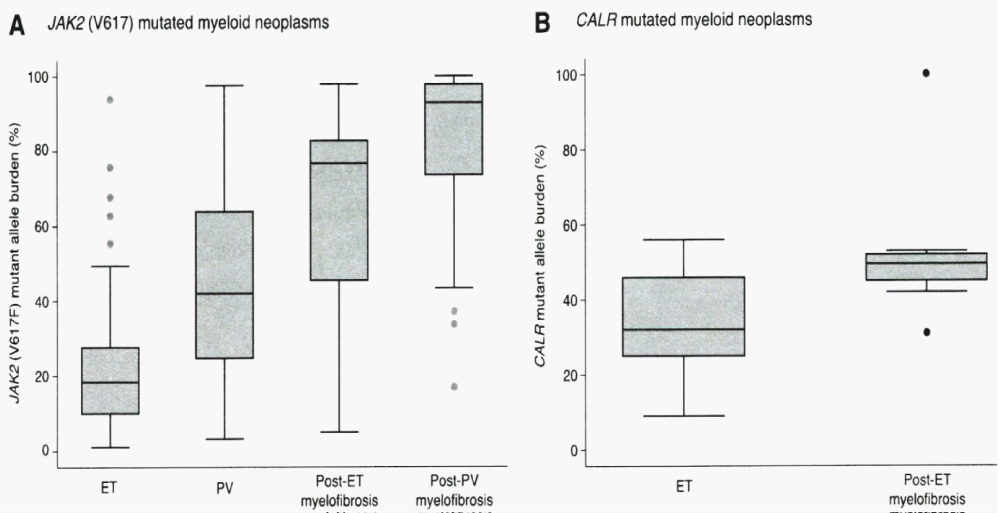
Figure 7: Granulocyte JAK2V617F mutated allele burden in WHO defined 250 ET, 212 PV, 18 post-Et and 55 post-PV patients from the study of Rumi. et al. [76]. JAK2V617F allele burden was recorded in granulocytes positivity above 50% (change from heterozygoushomozygous) in 2%, 41%, 72%, and 93% of ET, PV, post-ET and post-PV myelofibrosis patients. Granulocyte CALR mutation lallele load in granulocytes of selected 38 CALR ET patients at time of diagnosis was below 50% (median 32%) and around 50% in 10 CALR post-ET MF patients (median 50%) in the study of Rumi, et al. [76].
The UK MPN Study Group [71,78] elegantly confirmed the Vainchenker’s ‘dosage’ concept at the biological EEC level by studying the genotype of individual BFU-E in a crossectional cohort of 29 JAK2V617F mutated ET and 30 JAK2V617F mutated PV patients (Figure 8). The JAK2 mutation load was expressed as a percentage (%) of EEC colonies genotyped as homozygous (red), heterozygous (purple) or wild type [78] (Figure 8). All 29 JAK2V617F positive ET patients have heterozygous JAK2 mutated EEC colonies: 9 of them have a low percentage (< 10%) of homozygous JAK2 mutated colonies. Out of 30 JAK2V617F positive PV patients, 8 have heterozygous JAK2 mutated EEC, 13 have homozygous EEC colonies of more than 50% and 7 of less than 50%. Homozygous EEC colonies were absent or rare in heterozygous ET, but prevalent in JAK2V617F-positive PV [78] (Figure 6). These observations are completely in line with Vainchenker’s “dosage” concept (Figures 5,6, Table 1) [8,9,21,60,72]. Additional cytogenetic [79], genetic or epigenetic alterations in PV and MF patients are of huge prognostic significance [80-84]. The presence of epigenetic factors like TET2 or ASXL1 etc on top of the JAK2, MPL and CALR driver mutations of MPN is associated with impaired prognosis in MPN, MDS and other myeloid malignacies as well. The targeted search for epigenetic factors will become hugely important to the understanding of differences in biology, prognosis and outcome of MPN patients [81-84]. Using next generation sequencing (NGS) on top of the JAK2 or CALR mutation, the Swiss MPN investigators in Basel found one, two or more epigenetic somatic mutations in 65 (33%) of 197 WHO defined MPN patients (94 PV, 69 ET, 34 MF) [82]. Seventeen of 69 (25%) ET patients, 11 of 34 (32%) MF and none (0%) of 94 PV patients carried mutations in CALR. In addition to JAK2V617F and CALR, the most frequently observed epigenetic somatic mutations affecting the biology and natural history of MPN disease included TET2, ASXL1, DNMT3A, EZH2, and IDH1 [82-84]. Rare epigenetic mutations were NF1, NFE2, and RUNX1. The presence of one, two or more somatic mutations appeared to impair prognosis in JAK2 and CALR mutated MPN [82]. Tefferi, et al. [83,84], confirmed the Lundberg observations in large scale retrospective studies in WHO defined ET, PV and MF patients demonstrating that epigenetic somatic mutation detection on top of the JAK2, CALR and MPL mutational load and subtype MPN characteristization is far superior to classify the distinct MPN diseases as compared to the crude WHO classification, that cannot clearly distinct between ET, prodromal overt and masked PV and PV with MF. Dr. Green, addressed the key question whether the sequence of acquisition of somatic mutations can be inferred from the genotypes of detectable subclones [85]. For instance, if some tumor cells have JAK2V617F, and others from the same patient bear JAK2V617Fwith an additional somatic mutation, then this indicates that JAK2V617Fcame first. Genotyping individual hematopoietic colonies has shown that the order of acquisition of JAK2V617F, relative to mutations in TET2 or DNMT3A, influences subclonal composition within HSPCs and mature cell compartments, disease presentation, and clinical outcome.In JAK2-first patients, the HSC compartment is dominated by double-mutant cells, and such patients present at a younger age, often with PV. Conversely, in TET2-first patients, the HSC compartment is dominated by single mutant cells, and such patients present at an older age, usually with ET. JAK2-first patients had a greater likelihood of presenting with PV than with ET, had an increased risk of thrombosis, and an increased sensitivity of JAK2 mutant progenitors to ruxolitinib in vitro.
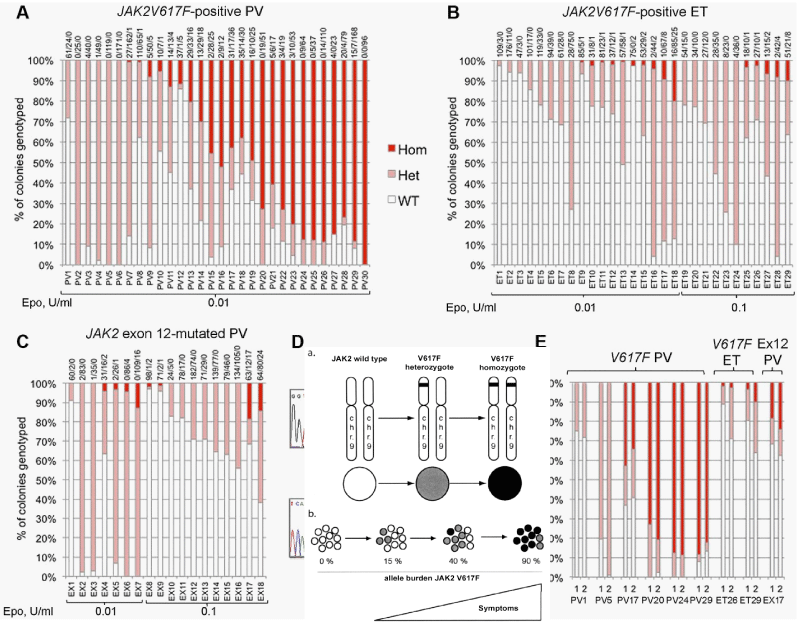
Figure 8: Proportions of JAK2 genotypes in BFU-Es from 59 patients with JAK2V617F-mutated essential thrombocythemia (ET) and polycythemia vera (PV) [78]. Each vertical bar represents 1 patient, divided according to the proportion of wild-type, heterozygous, and homozygous-mutant colonies obtained, with the absolute colony numbers shown above: (wild type white), heterozygous (purple) homozygous (red). Results of EEC colony genotypes are presented for 29 JAK2V617F-positive ET (B) patients (total 2277 colonies; mean 79 per patient) and for30 JAK2V617F-positive PV (A) patients (total 2287 colonies; mean 76 colonies per patient).All 29 JAK2V617F positive ET patients have heterozygous JAK2 mutated EEC colonies and less than 10% homozyous colonies in 9 and 20% in 1 of them. Out of 30 JAK2V617F positive PV patients 8 have heterozygous JAK2 mutated EEC, 13 have homozygous EEC colonies of more than 50% and 7 of less than 50%.
A. In total 29 PV patients: 5 were heterozygous, 13 heterozygous/homozygous and 11 predominant homozygous (high allele burden) for the JAK2V617F mutation.
In total 30 ET patients: all are predomominant heterozygous (low allele burden) for the JAK2V617F mutation but half of them do have a minor clone of homozygous mutated BFU-Es.
B. In total 18 JAK2exon 12 mutated PV: all are predominany heterozygous (low allele burden) for the JAK2exon 12 mutation, but 7 of them had a minor clone of homozygous mutated BFU-Es.
C and D. EEC colony genotypes for 18 patients with JAK2exon 12 mutated PV (total 1931 colonies; mean 107 per patient (C). D show example sequence traces for patients with patients with homozygous JAK2exon12 mutations in colonies. In total, 16 patients (5 “heterozygous-only” JAK2V617F-positive PV patients, 4 JAK2V617F positive PV patients with homozygous and heterozygous clones, 3 JAK2V617F positive ET patients with small homozygous clones, and 4 JAK2exon 12 mutated PV patients with homozygous clones) were assessed in this way (mean time between experiments, 13 months; range, 2-32 months) and showed reproducibility of proportions of heterozygous and homozygous-mutant colonies.
Interpretation. The JAK2V617F dosage concept of Constantinescu & Vainchenkeris in line with the EEC bone marrow findings in the UK study in Figures A and B [78]. A low level of JAK2V617F kinase activity only activate the MPL (TPO) receptor and favors the ET phenotype in acquired heterozygous (Figure 2, Table 1), [8,27,60,76] and in dominant heterozygous JAK2 or TPO mutated ET (Figure 2, HET), [28]. A high level of JAK2V617F activity in heterozygous/homozygous or homozygous mutated trilinear MPN is needed to activate the erythropoietin receptor (EPOR) and generate a PV-like phenotype (Figure 3), [8,21,31,32,57,59]. Similarly, high levels of JAK2V617F activity of long duration in homozygous mutated trilinear MPN is needed to activate the granulocyte clony-stimulating factor reeoptor (C-GCSF, Figure 3) leading to EMF or MG bone marrow phenotype and progressive secondary myelofibrosis (MF) [31,32,69,70]. Other mechanisms do occur in the pathophysiologies of myeloid metaplasia and myelofibrosis in advanced stage of trilinear MPNs.
Erythromelalgic microvascular circulation distubances or platelet thrombophilia in PV and ET: From Dameshek to Michiels & Van Vliet
Dameshek & Henthel, [1] described the presenting clinical manifestations in 20 newly diagnosed PV patients including quite severe headaches in 17, attacks of migraine in 14, visual disturbances, particularly spots before the eyes and coloured scotomas in 6, paresthesias numbing and tingling in toes and fingers in 12 and various types of transient major thrombosis (cerebral, coronary, venous) in 9 cases. Dameshek & Henthel, et al. [1] noted that the lack of large vessel involvement in PV and the associated high platelet counts suggested the possibility of ‘platelet thrombophilia’ as the cause of multiple small peripheral vascular thromboses in the peripheral cerebral and coronary circulation similar to aspirin-responsive platelet-dependent thrombophilia in JAK2V617F mutated thrombocythemia in ET and PV patients first discovered and described by Michiels Ten Kate & Van Vliet in [86] 1984 1985 and subsequently confirmed between 1985 and 2018 [20-22,35,36,86]. The broad spectrum of acroparesthesis, erythromelalgic and acrocyanotic ischemia or gangrene together with the episodic and transient neurlogic symptoms of migraine accompaniments, attacks of amnesia, dysbasia, dysphasia, TIA, hemiparesis and acute coronary ischemic syndromes all are the consequence of one underlying disorder: arterial thrombophilia causally linked to JAK2V617F platelet-mediated and MPL515platelet-mediated arteriolar inflammation and thrombosis in acquired thrombocythemias [20-22,27,28,35,36,86]. Platelet-mediated erythromelalgic microvascular disturbances also occur in dominant hereditary ET (HET) caused by heterozygous germ line gain of function mutation in the TPO, JAK2 and MPL genes [34,73-75] (Figure 5). Erythromelalgia is rare CALR mutated thrombocythemia and has never been observed in reactive thrombocytosis [35,36]. Platelet-mediated inflammatory and thrombotic processes in the end-arterial microcirculation typically respond to aspirin, but not to platelet ADP inhibitors and anticoagulation with vitamin K antagonists [20-22,35,36]. JAK2V617F mutated platelets are constitutively activated, hypersensitive (sticky) and cause aspirin responsive platelet-mediated microvascular circulation disturbances, (Table 1), [20,86-94] has recently been discovered by Michiels as the novel Aspirin-responsive Sticky Platelet Syndrome in JAK2V617F, MPL and TPO mutated thrombocythemias [35,36]./p>
JAK2exon12 mutations as cause of Isolated Erythrocythemia and PV
The finding of the JAK2exon12 mutations in the 5% PV patients negative for JAK2V617F usually present with early stage PV or isolated erythrocythemia (IE, Figure 8) with increased red cell mass but normal leukocytes and platelets and no palpable spleen [95-98]. The frequency of JAK2exon12 mutations among all PV patients is estimated around 3% [95,98]. JAK2 N542-E543del is the most frequent among the different reported exon 12 mutations. JAK2exon12 mutated MPN patients with increased erythrocytes above 6.0x1012/L and a typical PV bone marrow histology are diagnosed as benign IE or PV with a favourable outcome and normal life expectancy [95,96,98,99]. Pre-treatment bone marrow histology in JAK2exon 12 mutated PV or IE showed characteristic erythroid hyperplasia with minor and distinct histology changes of the megakaryocytic lineage, which are not seen in primary or secondary erythrocytoses (PE and SE) [95]. Cases of JAK2exon12 mutated IE or PV have erythrocytes above 6x1012/L [100], normal platelet and leukocyte counts, no or palpable spleen and a typical hypercellular bone histopathology predominantly due to erythroid hyperplasia and clusters of large megakaryocytes with hyperploid nuclei [95,98] (Figure 8). Bone marrow histology in 7 cases (4 IE, 3 PV) of JAK2exon12 mutated MPN in the pathology study of Lakey, et al. [97], showed prominent hyperplasia of erythropoiesis and atypical small to medium-sized large megakaryocytes (Figure 8). A low percentage of homozygosity was found for the JAK2 K539L-type and E543del-type exon 12 mutations (Figure 8) [78]. Godfrey, et al. [78], assesed the colony genotypes for 18 patients with JAK2exon 12-mutated PV in a total of 1931 colonies; mean 107 per patient (Figure 8C).
Example sequence traces for patients with patients with homozygous JAK2exon12 mutations in colonies are shown in figure 8D. In total, 16 patients (5 “heterozygous-only” JAK2V617F-positive PV patients, 4 JAK2V617F-positive PV patients with homozygous and heterozygous clones, 3 JAK2V617F-positive ET patients with small homozygous clones, and 4 JAK2exon12 mutated PV patients with homozygous clones showed reproducibility of proportions of heterozygous and homozygous-mutant colonies (Figure 8D).
Acquired MPL515 mutated normocellular ET
The prevalence of the MPL515 mutated ET range from 3% of MPN to 8.5% of JAK2 wild type ET and MF [101-103]. The clinical presentation in 30 MPL515 mutated ET patients (9 males and 21 females, age 22-84, mean 56 years) was featured major arterial thrombosis in 23%, venous thrombosis in 10%, aspirin responsive microvessel disturbances in 60%, and major hemorrhage in 7% [101]. The clinical, laboratory, molecular and pathological (CLMP) findings in MPL515 mutated ET were increased platelet count, 956+331×109/L in all, slight splenomegaly in 5 (17%), and no PV features in blood and bone marrow in all table 6, [31,32,34]. Pretreatment bone marrow histology at the time of diagnosis in MPL515 mutated ET features large and giant megakarocytes with hyperlobulated staghorn-like nuclei (Figure 10, Table 6), clearly different from JAK2exon 12 PV (Figure 9), and distinct from JAK2V617F ET and prodromal PV (Figures 2,3,9) and distinct from CALR thrombocythemia (Figures 11,12).
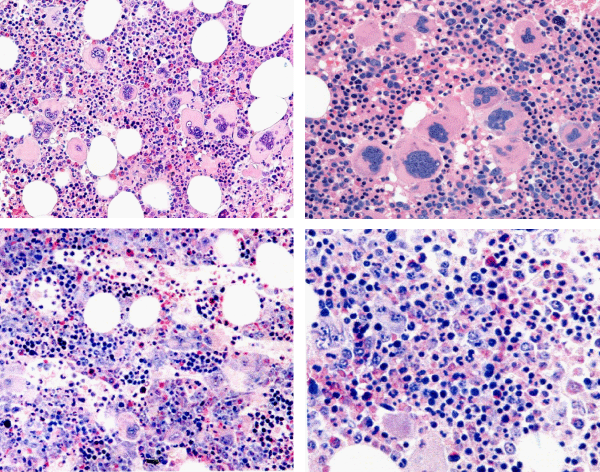
Figure 9: Upper left: Prodromal PV with hypercellular bone marrow (70%) due to increased erythropoiesis proliferation of mature medium to large pleomorphic megakaryocytes with variable degrees of hyperlobulated nuclei. Courtesy of Pathology Laboratory Pich. Upper right: Pre-treatment bone marrow histology in JAK2exon 12 mutated PV or E showed characteristic erythroid hyperplasia with distinct histology changes of the megakaryocyte lineage with no or minor lobulation of nuclei [97]. Lower left and right: Pleiomorphic small to large megakaryocytes with hyperlobulated nuclei in JAK2V617F positive ET (200x and right 400x Pich, et al. [66]).
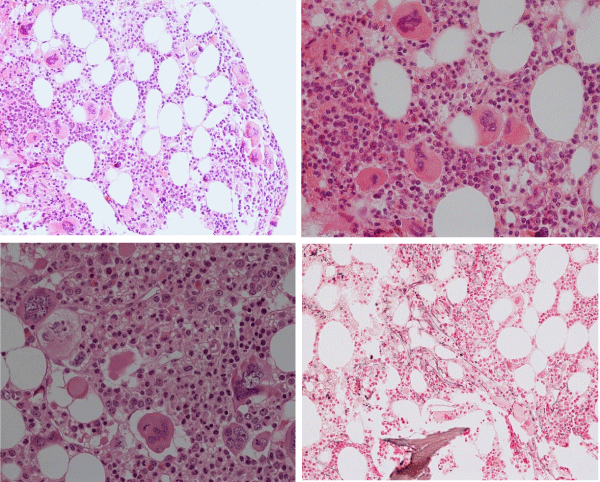
Figure 10: Dense clustering of mature, large to gaint megakaryocytes with hyperlobulated staghorn-like nuclei in a normocellular bone marrow of a symptomatic (aspirin responsive microvascular disturbances) case of MPL515 mutated thrombocythemia (platelet 1239x109/L) without features of PV in blood and bone marrow. (Pathology Laboratory Brussels, Dr De Raeve).
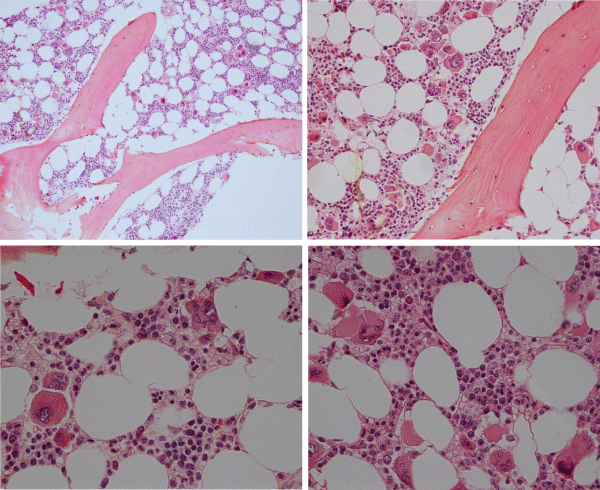
Figure 11: Loose dense clustering of large immature megakaryocytic (M) proliferation with immature ‘cloud’-like nuclei in a normocellular bonemarrow of an asymptomatic case of prefibrotic CALR thrombocythemia at platelet count of 879x109/L, minor splenomegaly on echogram and no features of PV in blood and bone marrow. (Pathology Laboratory Brussels, Dr De Raeve).
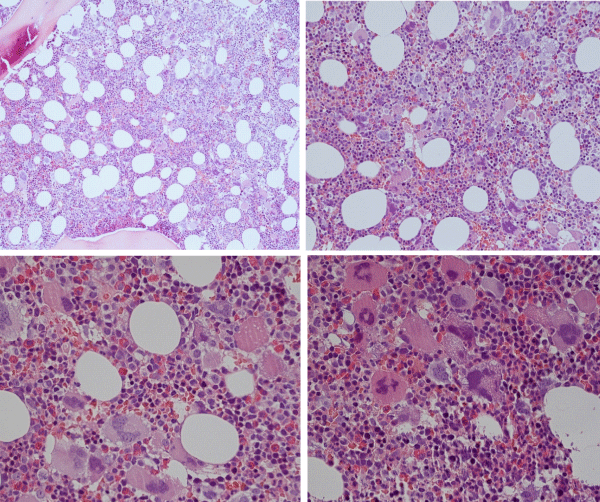
Figure 12: CALR mutated prefibrotic hypercellular ET or primary megakarocytic, granulocytic myeloproliferation (PMGM) with clustred increase of large immature megakaryocytes with immature ‘cloud-like’ nuclei diagnostic for prefibrotic CALR mutated thrombocythemia.
Megakaryocyte Leukemia (ML) and CALR mutated Thrombocythemia: From Dameshek 1951 to Kralovics 2013 and Michiels 2015
According to Dameshek, [10] mega karyocyte leukemia (ML) is defined by platelet counts around and above 1000x109/L without features of PV in blood and bone marrow smear and biopsy. The traditional classification of the myeloproliferative disorders (MPD) by the PVSG and used in textbooks was revised in the Hannover Bone Marrow classification to include PV, primary thrombocythemia (PTH), and hyper cellular thrombocythemia related to primary megakaryocytic myeloprliferation (PMGM, Table 7) without features of PV [11,12,14,16,104]. The discovery of the calreticulin (CALR) as the main cause of JAK2/MPL515 wild type thrombocythemia and PMF by Kralovics and his team [105] was identified by Michiels & De Raeve [31,32] as the driver cause of prefibrotic and fibrotic stages of PMGM without features of PV. This led to the second ground breaking event in the molecular landscape of the MPNs that induced a complete revision of all MPN classifications of the PVSG, WHO into the current Clinical Laboratory, Genetic and Pathobiological (2018 CLMP) criteria for JAK2V617Ftrilinear MPN (Tables 3 and 4), and JAK2exon 12 PV as compared to two distinct MPL515 (Table 6) and CALR thrombocythemias and myelofibrosis (Table 7) without features of PV.
Kralovics performed targeted whole-exome seqencing in 6 cases of WHO defined JAK2/MPL wild type PMF patients and found somatic calreticulin (CALR) mutations of 52-bp deletion in 1, of 1bp deletion in 1 and recurrent 5-bp insertion in 4 MF patients. The CALR somatic mutation was subsequently discovered as the driver cause of thrombocythemia in 78 of 311 (25%) ET patients and in 72 of 203 (35%) MF patients [105]. The CALR mutation was detected in none of 382 PV, 45 CML, 73 MDS, and 64 chronic myelomonocytic leukemia (CMML) patients. Three (12%) of 24 RARS-T cases were positive for both the SF3B1 and CALR mutation. A subsequent Italian-Austrian study of 1235 WHO-defined ET and MF patients detected the JAK2V617F, MPL515 and CALR mutation in 63.3%, 23.5% and 4.4% respectively with 8.8% being negative for all three mutations [76] (Figure 6). Evolution into MF during follow up was as high in CALR mutated ET as in JAK2V617F mutated PV (about 20% after 20 years). CALR mutated MPN patients lacked features of PV (normal erthrocytes and hematocrit), had higher platelet counts and a lower incidence of major thrombosis compared to JAK2V617F positive ET [76,105]. The large UK study confirmed the presence of the somatic CALR driver mutations in 80 of 112 (70%) JAK2/MPL wild type ET patients, and in 18 of 32 (56%) JAK2/MPL wild type MF patients and in none of 120 JAK2V617F or MPL515 mutated MPN patients [106]. CALR mutations were detected in 10 of 120 (8%) MDS patients (RA in 5 of 53, RARS in 3 of 27 and RAEB-T in 2 of 27), and in one patient each with CMML and atypical CML. CALR mutations were not found in control samples, lymphoid cancers, solid tumors, or cell lines [106]. A third large Italian study found CALR mutations in 15.5% of 576 WHO-defined ET and in 48.9% of JAK2/MPL wild type ET patients [107]. The distribution of the JAK2V617F, CALR and MPL515 mutations or triple negative cases in 254 WHO-defined MF patients was 58%, 25%, 8.3% and 8.7% with median overall survival of 8.2, 4.1, 4.3 and 2. 5 years respectively reflecting advanced or end stage MPN disease [39].
The biological and clinical features of WHO-defined ET carrying the JAK2V617F and CALR mutation ET clearly differ [76]. The mutant allele burden was lower in JAK2V617F mutated than in CALR mutated ET (Figure 7). JAK2V617F ET patients were older, had higher hemoglobin and white blood cell counts but lower platelet counts. Serum erythropoietin levels are lower and frequently decreased in JAK2V617F ET but normal in CALR thrombocythemia. The cumulative risk of WHO-defined ET carrying the JAK2V617F mutation to transform into WHO-defined PV was 29% after 15 years but transformation into PV was never observed in CALR thrombocythemia. With the advent of the CALR mutation as the main driver cause of JAK2/MPL wild type ET, hypercellular ET associated with PMGM [8,11,12,14,15] and CALR thrombocythemia and myelofibrosis appeared to be the same distinct MPN entity without features of PV (Table 7), [31-33]. JAK2V617F mutated ET and PV patients had a similar two times higher risk of major thrombosis than that of CALR mutated thrombocythemia patients. CALR-mutated ET patients were more frequently male, had higher platelet counts, lower hemoglobin and leukocyte count and showed a lower risk of major thrombosis than JAK2 mutated ET patients in two large studies [107,108].
Incorporation of 2008 WHO into 2020 clinical, laboratory, molecular and bone marrow pathology (CLMP) classification of myeloproliferative neoplasms
Bone marrow histology findings in 59 WHO-defined JAK2V617F positive ET and 44 JAK2 wild ET cases in the study of Pich, et al. [66], (Figure 11) revealed PV-like hypercellular morphological bone marrow changes of pleiomorphic enlarged megakaryocytes in JAK2V617F mutated ET similar as described previously (Figure 9), [8,15]. Various stages erythropoiesis and or myelopoiesis with megakaryocyte proliferation as well as LDH and spleen size are more pronounced in PV-like phenotype in JAK2V617F mutated ET in particular at higher JAK2 mutation load (Figure 9), [66]. WHO defined JAK2V617F positive ET showing increased cellularity due to increased erythropoiesis is consistent with prodromal PV [62]. The prognosis of JAK2V617F mutated ET and prodromal PV is favorable and to be treated with low aspirin and additional phlebotomy in early PV to maintain ht below 0.45 in man and below 0.42 in women. This concept based on prospective clinical observations are completely in line with the present study of patients with JAK2V617F mutated ET, prodromal PV and PV.
Bone marrow histology analysis of bone marrow biopsies by Michiels & De Raeve from the Vannucchi’s study on WHO defined MPL515 mutated ET revealed that clustered large to giant maure megakaryocyte with staghorn nuclei and platelet count increase in a normocellular bone marrow are characteristic for JAK2 wild type ET carrying the MPL515 mutation [31-33]. JAK2/CALR wild type ET carrying the MPL515 mutation indeed displayed clustered large and giant mature megakaryocytes with a greater number of large deeply lobulated ‘staghorn’ nuclei in a normocellular bone marrow as the hallmark of MPL515 thrombocythemia (Figure 10), [6,7,31,32].
Between 2015 and 2018 Michiels & De Raeve found typical PMGM pictures in 15 CLMP defined consecutive newly diagnosed CALR mutated ET (Figures 11,12) and MF patients [6,7,31-33]. CALR thrombocythemia patients appeared to be phenotypically identical to JAK2 wild type PMGM defined by the Hannover Bone Marrow Classification and in retrospect surely belong to the original description by Dameshek of megakaryocyte leukemia (ML) without features of PV [10]. CALR mutated thrombocythemia and MF are clearly distinct from MPL515 normocelluar thrombocythemia (Figure 10), JAK2V617F ET, prodromal PV and PV cases with regard to clinical, hematological and bone marrow features at presentation and during follow-up (Figure 9).
The European Asiatic collaboration between Michiels & De Raeve (Rotterdam-Brussels) and Yongoo and Myungshin Kim (Seoul, Korea) translated the laboratory, molecular and pathological characteristics in a large cross sectional study of 407 WHO defined MPN patients into the 2015-2020 CLMP classification (Tables 2-7). The Large cohort of 407 MPN patients included PV in 111 (29%), ET in 179 (44%) and PMF in 117 (29%). The three driver mutations were detected in 82.6% of 407 MPN patients with a mutation distribution of JAK2 in 275 (67.5%), CALR in 55 (13.7%), MPL in 6 (1.5%) [100]. In this report we analyzed the CLMP characteristics of 337 Korean evaluable WHO defined MPN patients subdivided into JAK2V617F in 268 (80%), JAK2exon 12 in 7 (2.1%, CALC in 56 (17%) and MPL in 6 (1.8%) [6,100]. The values of hemoglobin (Hb), hematocrit (Ht) and erythrocytes in JAK2V617F mutated trilinear MPN ranged from anemic to polycythemic values with mean values of Hb 14.7 g/dL, Ht 0.44 and erythrocytes 5.0x1012/L (Table 8). The bone marrow (BM) lineage proliferation class in MPN including 101 PV, 95 ET and 78 PMF WHO defined patients MPN consisted of M (WHO-ET) in 80; EM and EMG in 116 consistent with prodromal and classical PV; and GM myelofibrosis in 72. The mean JAK2V617F mutation load was high 69 to 80% in EM, EMG and 69% in MG bone marrow class, but low (37%) in M class ET patients (Table 8).
JAK2exon12 patients in the study of Kim, et al. [100], are featured by idiopathic erythrocythemia (IE) not meeting WHO-defined PV with normal platelet and leukocyte counts, no or palpable spleen and a hypercellular bone marrow predominantly due to erythroid hyperplasia (EM, Table 8) [71,95-98]. JAK2exon12 mutated MPN in the study of Kim et al presented with erythrocyte counts above 5.8x1012/L, normal platelet counts of less than 350x109/L and no anemia consistent with the diagnosis of erythrocythemic PV (Table 8). Increased erythropoiesis in bone marrow was absent in all cases of CALR and MPL mutated MPN (Table 8). Bone marrow histology in 56 cases of CALR mutated MPN typically featured predominant increased monolinear megakaryopoiesis M in two thirds and increased granulopoiesis and megakaryopiesis (GM) in one third (Table 8).
Table 8: Change of 2008/16 WHO into European Asiatic 2015-2020 CLMP characteristics in 337 patientswith Myeloproliferative Neoplasms (MPN) caused by the somatic driver mutations in the JAK2V617F, JAK2exon12, and CALR [100].
|
|||
| 337 MPN patients | JAK2V617F | JAK2exon12 | CALR |
| Patients N | 268 | 7 | 56 |
| % of 337 | 80% | 2.1% | 16.6% |
| Age yrs | 66 | 66 | 56 |
| Range | 22-89 | 46-76 | 20-89 |
| Males (%) | 45.5% | 28.6% | 41.1% |
| Hemoglobin g/dL | 14.7 | 18.3 | 12.6 |
| Range | 6.2-22.6 | 13.7-21.1 | 7.5-16.1 |
| Hematocrit (%) | 43.9 | 49.9 | 38.4 |
| Range | 19.7-69-1 | 46.2-59.3 | 22.9-47.0 |
| Red Blood cells | 5.01012/L | 6.91012/L | 4.2 1012/L |
| Range | 1.89-9.72 | 5.83-8.50 | 2.25-5.32 |
| Platelets 109/L | 650 | 281 | 898 |
| Range | 13-3268 | 58-310 | 49-1795 |
| Leukocytes 109/L | 12.0 | 8.2 | 8.6 |
| Range | 2.2-177 | 6.2-22 | 4.8-31 |
| 2008/16 WHO Class | JAK2V617F | ||
| PV N | 101 | 6 | 0 |
| ET N | 95 | 0 | 40 |
| PMF N | 78 | 1 | 16 |
| BM CLMP Class | |||
| EM N | 32 PV | 5 IE PV | 0 |
| EGM N | 84 PV | 2 PV | 0 |
| MET N | 80 | 0 | 37 |
| GMMF | 72 | 0 | 19 |
| Mutation burden | |||
| Total-Range | 67% (1.8-99) | 43.5% (34.5-49.4) | 48% (17.8-93) |
| EM + EGM | 85% (13-99) | 43.5% (34.5-49,4) | 0 |
| M (ET) | 37%ET | No data | *Type 1 53% |
| GM (MF) | 69%ET PV | No data | *Type 2 38% |
| Abbreviations: PV: Polycythemia Vera; ET: Essential Thrombocythemia; PMF: Primary Myelofibrosis; E: Erythroid; M: Megakaryocytic, G: Granulocytic myeloproliferation of increased bone marrow proliferation lineage. N: Number; Red = JAK2V617Fmutated PV, ET or MF. JAK2 wild type either CALR (blue) or MPL515 (black) mutated. *CALR type 1 revealed ahigher mutant alleleburden (53%) comparedto CALR type 2 (38%) MPN JAK2V617F mutated ‘forme fruste’ (prodromal PV) and early PV, and exon 12 PV patients presented with E(M) bone marrow proliferation without fibrosis. |
|||
The grade of bone marrow (BM) fibrosis in the study of Kim, et al. was divided into minimal fibrosis MF 0/1 and overt fibrosis MF 2/3 [7,11,12,21,109]. The frequency of overt fibrosis in JAK2V617F- and CALR-mutated and triple-negative MPN patients was 22.2%, 27.1% and 29.3%, respectively. JAK2-GM and CALR-GM showed a high rate of overt fibrosis (46.0 and 42.1%), followed by JAK2-M (17.5%), CALR-M (17.2%) and JAK2-EMG) (10.4%; p < 0.001). None of the JAK2-EM (‘forme fruste’, early and overt PV and exon 12 PV) patients presented overt fibrosis.
The overall bone marrow histology findings of erythroid, granulocytic and/or megakaryocytic hyperplasia in JAK2V617F mutated MPN, and of granulocytic and/or megakaryocytic hyperplasia in CALR mutated MPN patients in the Seoul study are completely in line with the 2015-2020 CLMP classification of six distinct MPN disease entities and transitional MPN states. Comparing the survival curves of 2008/2016 WHO defined PV, ET and PMF versus the 2015-2020 CLMP defined JAK2V617F, JAK2exon12, CALR and MPL515 defined MPN without fibrosis versus with fibrosis strongly suggest that bone marrow fibrosis (BMF) grade MF 0/1 versus grade 2/3 appeared to be main adverse prognostic factor when associated with JAK2V617F and triple negative MPN disease (Figures 13,14), [100].
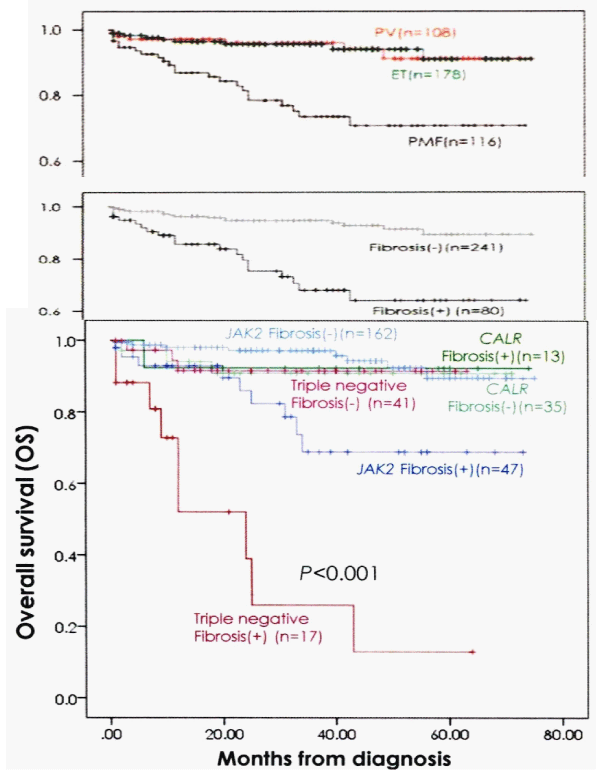
Figure 13: Comparing the survival curves of WHO defined PV, ET and PMF (upper curve) versus CLMP defined MPN without fibrosis (MF grade 0/1) versus with fibrosis (MF grade 2/3) in JAK2, CALR and triple negative MPN demonstrates that fibrosis is a measurable and objective adverse prognostic factor when associatedwith JAK2V617F and triple negative MPN disease (bottom). (Kim, et al. 2016) [100].
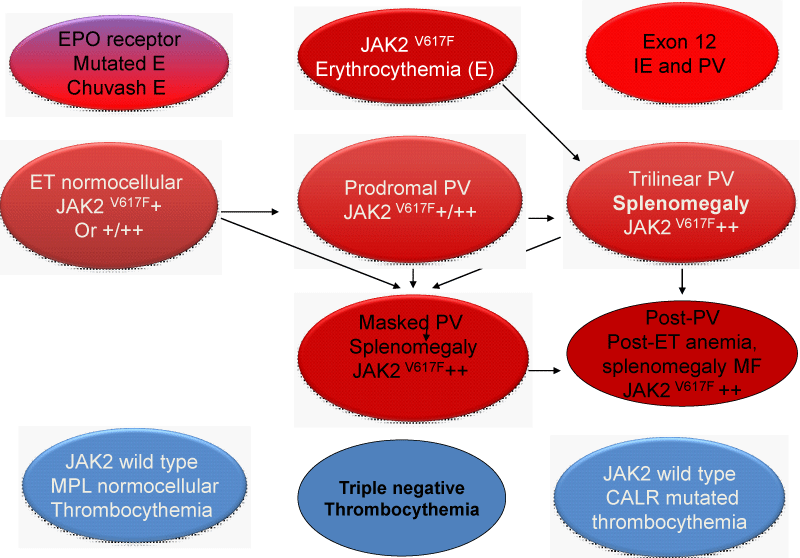
Figure 14: Clinical Laboratory Molecular and Pathobiology (2015-2020 CLMP) translational states of JAK2 mutated Myeloproliferative Neoplasms : MPN versus EPO receptor dominant congenital erythrocytosis caused by erythrocytosis (E). Transition of heterozygous into homozygous JAK2V617F mutation due to mitotic recombination is associated with a change of clinical ET phenotype into PV and MF phenotype of trilinear myeloproliferative neoplasm (MPN) during life long follow-up (Dameshek 1950, [2,57,58,60,21,31,32] James, et al. 2005, Kralovics, et al. 2005, Villeval,, et al. 2006, Michiels, et al. 2006, Bellucci & Michiels 2006, Michiels, et al. 2015ab). CALR or MPL515 thrombocythemia and bone marrow fibrosis (BMF) are distinct MPNs that mutually exclude each other and do not have PV features seen in JAK2V617F trilinear MPN and JAK2exon12 E or PV..
The present insight review is a strenuous joint effort by a multicentre MPN European Asiatic collaborative study group to demonstrate that scrutinized and integral clinical, laboratory, genetic and pathological (2015-2020 CLMP) approaches and intense communications amongst clinicians, scientist, molecular biologists, and pathologists are warranted to more precisely diagnose and treat each MPN patient before avoidable major complications had occurred. The change of 2008/2016 WHO into the 2015-2020 CLMP criteria in table 8 incorporate the established 1975 PVSG and 2001/2008/2016 WHO classifications. The novel 2015-2020 CLMP criteria for at least five distinct clonal MPNs are in urgent need of validation in well designed large clinical prospective unmet need (PUN) studies within the context of the International Collaborations and Academic Research on MPN (ICAR.MPN 2015 founded and chaired by Dr. Michiels Europe and Dr. Shuvaev, Russia) to even better define improved standards for diagnosis, classification, natural history and novel treatment options of JAK2V617F, JAK2exon12, CALR and MPL515 mutated myeloproliferative neoplasms [7,35,36,110,111-122].
The authors are gratefull to Alexander Georgii, Juergen Thiele, Ayalew Tefferi, Alessandro Vanucchi, Tiziano, Barbui, Stefan Constantinescu, William Vainchenker, Anthony Green, Radek Skoda, Robert Kralovics, Martin Griesshammer, Heinz Gisslinger, Hans Hasselbalch, Jiri Schwarz and Jean Jacques Kiladjian for their original and expert contributions to our current understanding of the JAK2V617F, JAK2Exon 12, CALR and MPL515 mutated myeloproliferative neoplasms (MPNs).
- Dameshek W, Henstell HH. The diagnosis of polycythemia. Ann Intern Med. 1940; 13: 1360-1387.
- Dameshek W. Physiopathology and course of polycythemia vera as related to therapy. JAMA. 1950; 142: 790-797. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15405984
- Videbak A. Polycythemia vera. Course and prognosis. Acta Med Scand. CXXXVIII. 179-197.
- Dameshek W. The treatment of Polycythemia. Blood. 1946; 1: 256.
- Michiels JJ. Physiopathology, etiologic factors, diagnosis and course of polycythemia vera as related to therapy according to William Dameshek 1940-1950. Turk J Hematol. 2013; 30: 102-110. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24385771
- Michiels JJ, De Rave H, Valster F, Potters V, Kim Y, et al. Extension of 2016 World Health Organization (WHO) classification and a new set of clinical, laboratory, molecular and pathological criteria for the diagnosis of myeloproliferative neoplasms: from Dameshek to Vainchenker, Green and Kralovics. EMJ. 2017a; 2: 72-81.
- Michiels JJ, Berneman Z, Gadisseur A, Raeve HD, Schroyens W, et al. Myelofibrosis is a Secondary Event in JAK2 Trilinear Myeloproliferative Neoplasm (MPN) and in CALR and MPL Thrombocythemia: Implications for Novel Treatment Options of Prefibrotic MPN. J Hematol Thromembolic Dis. 2017b; 5: 5.
- Michiels JJ, Hendrik De Raeve, Berneman Z, Van Bockstaele D, Hebeda K, et al. The 2001 world health organization and updated european clinical and pathological criteria for the diagnosis classification and staging of the Philadelphia-negative chronic myeloproliferative disorders. Sem Thromb Hemost. 2006a; 32: 307-340.
- Michiels JJ, Berneman Z, Van Bockstaele D, Van Der Planken M, De Raeve H, et al. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Sem Thromb Hemost. 2006b; 32: 174-207. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16673274
- Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951; 6: 372-375. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14820991
- Georgii A, Vykoupil KF, Buhr T, Choritz H, Döhler U, et al. Chronic myeloproliferative disorders in bone marrow biopsies. Path Res Pract. 1990; 186: 3-27.
- Georgii A, Buhr T, Buesche G, Kreft A, Choritz H. Classification and staging of Ph-negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leuk Lymphoma. 1996; 22: 15-29. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8951769
- Michiels JJ, Prins MEF, Hagemeijer A, Brederoo, P, van der Meulen J, et al. Philedelphia chromosome positive essential thrombocythemia and megakaryoblast leukemia. Am J Clin Pathol. 1987; 88: 645-752. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3479003
- Michiels JJ. Diagnostic criteria of the myeloproliferative disorders (MPD): essential thrombocythemia (ET), polycythemia vera (PV) and chronic megakaryocytic granulocytic metaplasia (CMGM). Neth J Med. 1997; 51: 57-64. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9286142
- Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemiavera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol. 2002; 76: 133-145. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12215011
- Michiels JJ, Kutti J, Stark P, Bazzan M, Gugliotta L, et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thromboythemia, polycythemiavera and essential megakaryocytic granulocytic myeloproliferation and myelofibrosis. Neth J Med. 1999; 54: 46-62. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10079679
- Michiels JJ, Juvonen E. Proposal for revised diagnostic criteria of essential thrombocythemia and polycythemia vera by the Thrombocythemia Vera Study Group. Sem Thromb Hemost. 1997; 23: 339-347. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9263350
- Kurnick JE, Ward HP, Block MH. Bone marrow sections in the differential diagnosis of polycythemia. Arch Pathol. 1972; 94: 489-499. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/5086062
- Ellis JT, Silver RT, Coleman M, Geller SA. The bone marrow in polycythemia vera. Sem Hematol. 1975; 12: 433-444. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/1198128
- Michiels JJ, Abels J, Steketee J, van Vliet HH, Vuzebvski VD. Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med. 1985; 102: 466-471. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3977194
- Bellucci S, Michiels JJ. The role of JAK2V617F mutation, spontaneous eryhtropoiesis and megakaryopoiesis, hypersensitive platelets, activated leukocytes, and endothelial cells in the etiology of thrombotic manifestations in polycythemia vera and essential thrombocythemia. Sem Thromb Hemost. 2006; 32: 381-398.
- Michiels JJ. Platelet-dependent and aspirin-responsive arterial thrombophilia in essential thrombocythemia. The Thoraxcentre J. 1996; 8: 1-4.
- Michiels JJ. Bone marrow histopathology and biological markers as specific clues to the differential diagnosis of essential thrombocythemia, polycythemiavera and prefibrotic or fibrotic myeloid metaplasia. Hematol J. 2004; 5: 93-102. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15048058
- Michiels JJ, De Raeve H, Hebeda K, Lam KH, Bot F, et al. Biology Diagnosis and Classification of MPD. Furst International Lymphoma-Leukemia-Myeloma (LLM) Congress. Turk J Hematol. 2007; 24: 37-53.
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H. PVSG and WHO vs European Clinical, Molecular and Pathological (ECMP) criteria for prefibrotic myeloproliferative neoplasms. World J Hematol. 2013b; 2: 71-88.
- Michiels JJ, Ten Kate FWJ, Koudstaal PJ, Van Genderen PJJ. Aspirin responsive platelet thrombophilia in essential thrombocythemia and polycythemiavera. World J Hematol. 2013c; 2: 20-43.
- Michiels JJ, Ten Kate F, Lam KH, Schroyens W, Berneman Z, et al. The European clinical, Molecular and Pathological (ECMP) criteria and the 2007/2008 revision of the World Health Organization for the diagnosis, classification and staging of prefibrotic myeloproliferative neoplasms carrying the JAK2V617F mutation. Turk J Hematol. 2014a; 31: 239-254.
- Michiels JJ, Stasko J, Kubish P, Pich A, De Raeve H. Autosomal dominant hereditary essential thrombocythemia due to a gain of function mutation in the thrombopoietin (TPO) of JAK2 gene as the cause of dominant congenital aspirin sticky platelet syndrome. J Hematol Thromb Dis. 2014b; 2: 6.
- Michiels JJ, Pich A, De Raeve H, Campr V, Schwarz J. WHO clinical molecular and pathological (WHO-CMP) features of congenital MPLS505N and the acquired MPLW515L/K mutated essential thrombocythemia and myelofibrosis. J Hematol Thromb Dis. 2014c; 2: 6.
- Michiels JJ. Myeloproliferative and thrombotic burden and treatment outcome in thrombocthemia and polycythemia patients. World J Crit Care Med. 2015; 4: 230-239. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26261774
- Michiels JJ, Berneman Z,Schroyens W, De Raeve H. Changing concepts of diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms: From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol. 2015a; 133: 71-86. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25116092
- Michiels JJ, Valster F, Wielenga J, Schelfout K, De Raeve H. European vs 2015 World Health Organization clinical molecular and pathological classification of myeloproliferative neoplasms. World J Hematol. 2015b; 4: 16-53.
- Michiels JJ, Medinger M De Raeve H, Schroyens W, Schelfout K, Potters V, et al. Increased erythrocyte count on top of bone marrow histology, but not by EPO level or JAK2V617F mutation load discriminates between JAK2V617F mutated essential thrombocythemia and polycythemia vera. J Hematol Thromb Dis. 2015c; 001.
- Michiels JJ, Tevet M, Trifa A, Niculescu-Mizil E, Lupa A, et al. 2016 WHO Clinical Molecular and Pathological Criteria for Classification and Staging of Myeloproliferative Neoplasms (MPN) Caused by MPN Driver Mutations in the JAK2, MPL and CALR Genes in the Context of New 2016 WHO Classification: Prognostic and Therapeutic Implications. MAEDICA. 2016; 11: 5-25. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28465746
- Michiels JJ. Aspirin-responsive eythromelalgia in JAK2-thrombocythemia and incurable inherited erythrothermalgia in neuropathic Naav1.7 sodium channellopathy: from Mitchel 1878 to Michiels 2017. Exp Opinon Orphan Drug. 2017c.
- Michiels JJ. Aspirin cures erythromelalgia and cerebrovascular distubances in JAK2-thrombocythemia. World J Hematol. 2017d; 6: 32-54.
- Wasserman LR. The management of polycythaemia vera. Br J Haematol. 1971; 21: 371-376. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/4941523
- Berlin NI. Diagnosis and classification of the polycythemias. Sem Hematol. 1975; 12: 339-351.
- Tefferi A, Pardanani A. Mutation screening for JAK2V617F: when to order the test and how to interpret the results. Leuk Res. 2006; 108: 3472-3476. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16460800
- Tefferi A, Pardanani A. Mutation screening for JAK2V617F: when to order the test and how to interpret the results. Leuk Res. 2006; 108: 3472-3476. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16460800
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127: 2391-2405. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31659364
- Michiels JJ. The Myeloproliferative Disorders. An historical appraisal and personal experiences. Leuk Lymphoma. 1996; 22: 1-14. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8951768
- Messinezy M, WestwoodNB, Woodstock SP, Strong RM, Pearson TC. Low seru, erythropoietin: a strong diagnostic criterion of primary polycythemia even et normal hemoglobin levels. Clin Lab Haematol. 1995; 17: 217-220.
- Messinezy M, Westwood NB, El-Hemaida I, Marsden JT, Sherwood RS, et al. Serum erythrpoietin values in erythrocytoses and in primary thrombocythemia. Br J Haematol. 2002; 117: 47-53. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11918532
- 2008 WHO criteria for polycthemia vera, primary myelofibrosis and essential thrombocythemia. Thiele et al In: Swerdlow SH, Campo E, Harris NL et al: WHO Classification of Tumours of Haematopoietic and Lympoid Tissues. Lyon France IARC. 2008; 40-50.
- Thomas DJ, du Boulay GH, Marshall J, Pearson TC, Ross Russell RW, et al. Cerebral blood-flow in polycythaemia. Lancet.1977; 2: 161-163. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/69781
- Thomas DJ, Marshall J, Russell RW, Wetherley-Mein G, du Boulay GH, et al. Effect of haematocrit on cerebral blood-flow in man. Lancet. 1977; 2: 941-943. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/72286
- Pearson TC, Wetherly-Mein. Vascular occlusive episodes and venous haematocrit in primary proliferative polycythaemia. Lancet. 1978; 2: 1219-1222. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/82733
- Messinezy M, Pearson TC, Prochazka A, Wetherley-Mein G. Treatment of primary proliferative polycythaemia by venesection and low dose busulphan: retrospective study from one centre. Br J Haematol. 1985; 61: 657-666. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/4084455
- Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med.2004; 350: 114-124. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14711910
- Marchioli R, Finazzi G, Vannucchi AM, Barbui T for the CTYO-PV Collaborative Group. Cardiovascular events and intensivity of treatment in polycythemia vera. N J Eng Med. 2013; 368: 22-33.
- Van Genderen PJJ, Michiels JJ. Hydroxyurea in essential thrombocytosis. N Engl J Med. 1995; 333: 802-803. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7643898
- Lengfelder E, Merx K, Hehlmann R. Diagnosis and therapy of polycythemia vera. Sem Thromb Hemost. 2006; 32: 267-275. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16673281
- Prchal JF, Axelrad AA. Bone marrow responses in polycythemia vera. N Eng J Med. 1974; 290: 1382. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/4827655
- Westwood NB, Person TC. Diagnostic applications of haematopietic progenitor culture tehniques in polycythaemias andthrombocythaemias. Leuke Lymphoma. 1996; 22: 95-103. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8951779
- Kralovics R, Guan Y, Prchal JT. Acquired uniparetal disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002; 30: 229-236. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11882360
- James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, et al. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythemia vera. Nature. 2005; 434: 1144-1148. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15793561
- Kralovics R, Passamonti F, Buser AS, Teo SS, Teidt R, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Eng J Med. 2005; 352: 1779-1790. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15858187
- Vainchenker W, Constantinescu SN. A unique activating mutation in JAK2 V617F is at the origin of polycythemia vera and allows a new classification of myeloproliferative diseases. Hematology Am Soc Hematol Educ Program. 2005; 195-200. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16304380
- Villeval JL, James C, Pisani DF, Casadevall N, Vainchenker W. New insights into the pathogenesis of JAK2V617F-positive myeloproliferative disorders and consequences for the management of patients. Sem Thromb Hemost. 2006; 32: 341-351.
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton et al. Acquired mutation of the tyrosine kinase in human myeloproliferative disorders. Lancet. 2005; 365: 1054-1061. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15781101
- Campbell PJ, Baxter EJ, Beer PhA, Scott LM, Bench AJ, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and the role in leukemic transformation. Blood. 2006; 18: 3548-3555. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16873677
- Moliterno AR, Williams DM, Isaacs MA, Spivak JL. Phenotypic variability within the JAK2V617F-positive MPD: roles of progenitor cell and neutrophil allele burden. Exp Hematol. 2008; 36: 1480-1486. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18723264
- Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OH. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011; 118: 1723-1735. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21653328
- Vainchenker W, Kralovics R. Genetic basis and molecular pathogenesis of classical myeloproliferative neoplasms. Blood. 2017; 129: 667-679. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28028029
- Pich A, Riera L, Beggiato E, Nicolino B, Godio L, et al. JAK2V617F mutation and allele burden are associated with distinct clinical and morphological subtypes in patients with essential thrombocythemia. J Clin Pathol. 2012; 65: 953-954. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22718845
- Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia. 2005; 19: 1847-1849. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16079890
- Gale RE, Allen AJR, Nash MJ, Linch DC. Log-term serial analysis of X-chromosome inactivation patterns andJAK2 V617F mutant levels in patients with essential thrombocythemia show that minor mutant-positive clones can remain stable for many years. Blood. 2007; 109: 1241-1243. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17023581
- Vannucchi AM, Antonioli E, Guglielmelli P, Longo G, Pancrazzi A, et al. Prospective identification of high-risk polycythemia vera patients based on JAK2V617F allele burden. Leukemia. 2007; 21: 1952-1959. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17625606
- Passamonti F, Rumi E, Pietra D, Della Porta MG, Boveri E, et al. Relation between JAK2V617F mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006; 107: 3676-3682. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16373657
- Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F JAK2 mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006; 108: 2435-2437. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16772604
- Delhommeau F, Pisani DF, James C, Casadevall N, Constatinescu S, et al. Oncogenic mechanism in myeloproliferative disorders. Cell Mol Life Sci. 2006; 63: 2939-2953.
- Mead AJ, Rugless MJ, Jacobsen SE, Schuh A. Germline JAK2 mutationin a family with hereditary thrombocytosis. N Eng J Med. 2012; 366: 967-969. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22397670
- Mead AJ, Chowdhury O, Pecquet C, Dusa A, Woll P, et al. Impact of isolatedgermline JAK2V617I mutation on human hematopoiesis. Blood. 2013; 121: 4156-4165. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23535062
- Etheridge SL, Cosgrove ME, Sangkhae V, Corbo LM, Roh ME, et al. A novel activating, germ line JAK2 mutation, JAK2R564Q, causes familialessential thrombocytosis. Blood. 2014; 123: 1059-1068. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24381227
- Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, et al. Jak2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcome. Blood. 2014; 123: 1552-1515. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24366362
- Lamy T, Devillers A, Bernard M, Moisan A, Grulois I, et al. In apparent polycythemia vera: an unrecognized diagnosis. Am J Med. 1997; 102: 14-20. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9209196
- Godfrey AL, Chen E, Pagano F, Ortmann CA, Silber Y, et al. JAK2V617F homozygosity arises commonly and recurrently in PV and ET, but PV is characterized by expansion of a dominant homozygous subclone. Blood. 2012; 120: 2704-2707. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22898600
- Bench AJ, Nacheva EP, Champion KM, Green AR. Molecular genetics and cytogenetics of myeloproliferative disorders. Baillieres Clin Haematol. 1998; 11: 819-848. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10640219
- Vannucchi A, Lasho TL, Guglielmelli P, Biamonte F, Pardani A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013; 27: 1861-1869. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23619563
- Guglielmelli P, Lasho TL, Rotunno G, JScore J, Mannarelli C, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014; 28: 1494-1500. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24549259
- Lundberg P, Karov A, Nienbold R, Looser R, Hao-Shen H, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014; 123: 2220-2228. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24478400
- Tefferi A, Laslo TL, Guglielmelli P, Finke CM, Rotunno G, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016; 1: 21-30. PubMed: https://pubmed.ncbi.nlm.nih.gov/29296692
- Tefferi A, Laslo TL, Finke CM, Elala Y, Hanson CA, et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016; 1: 105-111. PubMed: https://pubmed.ncbi.nlm.nih.gov/29296803
- Ortmann CA, Kent DG, Nangalia J, SilberY, Wedge DC, et al. Effect of mutation order on myeloproliferative neoplasms. N Eng J Med. 1015; 372: 601-612. PubMed: https://pubmed.ncbi.nlm.nih.gov/25946289/
- Michiels JJ, Ten Kate FWJ, Vuzevski VD, Abels J. Histopathology of erythromelalgia in thrombocythemia. Histopathology. 1984; 8: 669-678. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9263352
- Michiels JJ, Berneman Z, Schroyens W, Koudstaal PJ, Lindemans J, et al. Platelet-mediated thrombotic complications in patients with ET: reversal by aspirin, platelet reduction, and not by Coumadin. Blood Cells Mol Dis. 2006c; 36: 199-205. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16510297
- Michiels JJ, Berneman Z, Schroyens W, Koudstaal PJ, Lindemans J, et al. Platelet-mediated erythromelalgic, cerebral, ocular, and coronary microvasculae ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemiavera: a distinct aspirin-responsive and coumadin-resitent arterial thrombophilia. Platelets. 2006d; 17: 528-544. PubMed: https://pubmed.ncbi.nlm.nih.gov/17127481/
- Van Genderen PJJ, Michiels JJ, Van Strik R, Lindemans J, Van Vliet HHDM. Platelet consumption in thrombocythemia complicated by erythromelalgia: reversal by aspirin. Thromb Haemost. 1995; 73: 210-214. PubMed: https://pubmed.ncbi.nlm.nih.gov/7792731/
- Van Genderen PJJ, Lucas IS, Van Strik R, Vuzevski VD, Prins FJ, et al. Erythromelalgia in essential thrombocythemia is characterized by platelet activation and endothelial cell damage but not by thrombin generation. Thromb Haemost. 1996; 76: 333-338. PubMed: https://pubmed.ncbi.nlm.nih.gov/8883266/
- Van Genderen PJJ, Michiels JJ. Erythromelalgia: a pathognomonic microvascular thrombotic complication in essential thrombocythemia and polycythemia vera. Sem Thromb Hemost. 1997; 23: 357-363. PubMed: https://pubmed.ncbi.nlm.nih.gov/9263352/
- Van Genderen PJJ, Mulder PGH, Waleboer M, Moesdijk D, Michiels JJ. Prevention and treatment of thrombotic comlications in essential thrombocythemia: efficacy and safety of aspirin. Brit J Haematol. 1997a; 97: 179-184. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9136963
- Van Genderen PJJ, Leenknegt H, Michiels JJ. The paradox of bleeding and thrombosis in thrombocythemia: is von Willebrand factor the link? Sem Thromb Hemost. 1997b; 23: 385-389. PubMed: https://pubmed.ncbi.nlm.nih.gov/16977569/
- Van Genderen PJJ, Prins F, Michiels JJ, Schroer K. Thromboxane-dependent platelet activation in vivo precedes arterial thrombosis in thrombocythemia: A rationale for the use of low-dose aspirin as an antithrombotic agent. Br J Haematol. 1999; 104: 438-441. PubMed: https://pubmed.ncbi.nlm.nih.gov/10086775/
- Scott LM. TheJAK2 exon 12 mutants, a comprehensive review. Amer J Hematol. 2011; 86: 668-676. PubMed: https://pubmed.ncbi.nlm.nih.gov/21674578/
- Pardani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia. 2007; 21: 1960-1963. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17597810
- Lakey MA, Pardani A, Hoyer JD, Nguyen PL, Lasho TL, et al. Bone marrow morphologic features in polycythemia vera with JAK2 exon 12 mutations. Am J Clin Pathol. 2010; 133: 942-948. PubMed: https://pubmed.ncbi.nlm.nih.gov/20472853/
- Passamonti F, Elena C, Schnittger S, Skoda R, Anthony R Green et al. Molecular and clinical features of the myeloproliferative neoplasms associated with JAK2 exon 12 mutations. Blood. 2011; 117: 2813-2816. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21224469
- Scott LM, Tong W, Levine RL, Scott MA, Beer PA, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Eng J Med.2007; 356: 459-468. PubMed: https://pubmed.ncbi.nlm.nih.gov/17267906/
- Kim Y, Park J, Jo I, Lee GD, Kim J, et al. Genetic-pathologic characterization of myeloproiferative neoplasms. Exp Mol Med. 2016; 48: 247. PubMed: https://pubmed.ncbi.nlm.nih.gov/27444979
- Vannucchi AM, Antonioli E, Guglielmelli P, Pancrazzi A, Guerini V, et al. Charateristics and clinical correlates of MPL515W>L/K mutation in essential thrombocythemia. Blood. 2008; 112: 844-847. PubMed: https://pubmed.ncbi.nlm.nih.gov/18519816/
- Beer PA, Campbell PJ, Scott LM. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008; 112: 141-149. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18451306
- Jones AV, Campbell PJ, Beer PA, Schnittger, Vannucchi AM, et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood. 2010; 115: 4517-4523. PubMed: https://pubmed.ncbi.nlm.nih.gov/20304805/
- Michiels JJ, Berneman Z, Schroyens W, KuttiJ, Swolin B, et al. Philadelphia (Ph) chromosome positive thrombocythemia without features of chronic myeloid leukemia in peripheral blood and bone marrow: natural history and diagnostic differentiation from Ph-negative essential thrombocythemia. Ann Hematol. 2004; 83: 504-512. PubMed: https://pubmed.ncbi.nlm.nih.gov/15164229/
- Klampf T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, et al. Somatic mutations od calreticulin in myeloproliferative neoplasms. N Eng J Med. 2013; 369: 2379-2387. PubMed: https://pubmed.ncbi.nlm.nih.gov/24325356/
- Nangalia J, Massie CE, Baxter J, Nice FL, Gundem G, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014; 28: 1472-1477. PubMed: https://pubmed.ncbi.nlm.nih.gov/24402162/
- Rotunno G, Mannarelli C, Guglielmielli P, Pacilli A, Pancrazzi A, et al. Impact of calreticulin mutations on clinical and haematological phenotype and outcome in essential thrombocthemia. Blood. 2014; 123: 1552-1555. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24371211
- Andrikovics H, Krahling T, Balassa K, Halm G, Bors A, et al. Distinct clinical characteristics of myeloproliferative neoplasms with calreticulin mutations. Haematologica. 2014; 99: 1184-1190. PubMed: https://pubmed.ncbi.nlm.nih.gov/24895336/
- Thiele J, Kvasnicka HM, Facchetti F, Franco V, Van Der Walt J Orazi A. European consensus for grading bone marrow fibrosis and assessment of cellularity in myeloproliferative disorders. Haematologica. 2005; 90: 1128-1132. PubMed: https://pubmed.ncbi.nlm.nih.gov/16079113/
- Kiladjian JJ, Cassinat B, Turlure P, Cambier N, Roussel M, et al. High molecular response rate of polycythemia vera treated with peglyated interpheron-alpha-2a. Blood. 2006; 108: 2037-2040. PubMed: https://pubmed.ncbi.nlm.nih.gov/16709929/
- Larssen TS, Moeller MB, de Striker K, Peter Nørgaard, Jan Samuelsson, et al. Minimal residual disease and normalization of the bone marrow after longterm treatment with alfa-interferon2b in polycythemia vera. A report on molecular responses in seven patients in sustained complete hematological remission. Hematology. 2009; 14: 331-334. PubMed: https://pubmed.ncbi.nlm.nih.gov/19941739/
- Verger E, Cassinat B, Chauveau A, Dosquet C, Giradier S, et al. Clinical and moelucular response to interferon-alpha therapy in essential thrombocythemia patients with CALR mutations. Blood. 2015; 126: 2585-2691. PubMed: https://pubmed.ncbi.nlm.nih.gov/26486786/
- Wilkins BS, Erber WN, Bareford D, Buck G, Wheatley K, et al. Bone marrow pathology in essential thrombocythemia: interobserver reliability and utility for identifying disease subtypes. Blood. 2008; 111: 60-70. PubMed: https://pubmed.ncbi.nlm.nih.gov/17885079/
- James C, Delhommeau F, Marzac C, Teyssandier I, Le Couédic JP, et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia. 2006; 20: 350-353. PubMed: https://pubmed.ncbi.nlm.nih.gov/16341032/
- Cassinat B, verger E, Kiladjian JJ. Interferon alpha therapy in CALR-mutated essential thrombocythemia. N Eng J Med. 2014; 371: 188-189. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25006741
- Juvonen E, Ikkala E, Fyrquist F, Ruutu T. Autosomal dominant erythrocytosis caused by increased sensitivity to erythropoietin. Blood. 1991; 78: 3066-3069. PubMed: https://pubmed.ncbi.nlm.nih.gov/1954391/
- De La Chapelle A, Traskelin AL, Juvonen E. Truncated erythropietin receptor causes doinantly inheritedbenign erythrocytosis. Proc Natl Acad Sci USA. 1993; 90: 4495-4499. PubMed: https://pubmed.ncbi.nlm.nih.gov/8506290/
- Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006; 108: 1652-1660. PubMed: https://pubmed.ncbi.nlm.nih.gov/16670266/
- Laszlo J. Myeloproliferative disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD and hemorrhagic thrombocythemia. Semin Hematol. 1975; 12: 409-432. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/1105793
- Quintas-Cardama A, Kantarjian H, Manshouri T, Rajyalakshmi Luthra, Zeev Estrov, et al. Peglyated interferon alfa-2a yields high rates of hematological and molecular response in patients with advanced essential thrombocthemia and polycythemia vera. J Clin Oncol. 2009; 27: 5418-5424. PubMed: https://pubmed.ncbi.nlm.nih.gov/19826111/
- Vannucchi AM, Papaemmanuil E, Campbell PJ, and Green AR. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Eng J Med. 2013; 369: 2391-2405. PubMed: https://pubmed.ncbi.nlm.nih.gov/24325359/
- 2001 WHO classification of the chronic myeloproliferative diseases (CMPD) polycythemia vera, chronic idiopathic myelofibrosis essential thrombocythemia and cMPD unclassifiable. In: Jaffe SS, Harris NL, Stern A, Vardiman JW eds. WHO classification of Tumours of haematopoiesis and lymphoid tissues. Lyon, France IARC; 2001; 31-42.